High-Throughput Retroviral Tagging for Identification of Genes Involved in Initiation and Progression of Mouse Splenic Marginal Zone Lymphomas
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Genome-Wide Analysis of 5-Hmc in the Peripheral Blood of Systemic Lupus Erythematosus Patients Using an Hmedip-Chip
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 35: 1467-1479, 2015 Genome-wide analysis of 5-hmC in the peripheral blood of systemic lupus erythematosus patients using an hMeDIP-chip WEIGUO SUI1*, QIUPEI TAN1*, MING YANG1, QIANG YAN1, HUA LIN1, MINGLIN OU1, WEN XUE1, JIEJING CHEN1, TONGXIANG ZOU1, HUANYUN JING1, LI GUO1, CUIHUI CAO1, YUFENG SUN1, ZHENZHEN CUI1 and YONG DAI2 1Guangxi Key Laboratory of Metabolic Diseases Research, Central Laboratory of Guilin 181st Hospital, Guilin, Guangxi 541002; 2Clinical Medical Research Center, the Second Clinical Medical College of Jinan University (Shenzhen People's Hospital), Shenzhen, Guangdong 518020, P.R. China Received July 9, 2014; Accepted February 27, 2015 DOI: 10.3892/ijmm.2015.2149 Abstract. Systemic lupus erythematosus (SLE) is a chronic, Introduction potentially fatal systemic autoimmune disease characterized by the production of autoantibodies against a wide range Systemic lupus erythematosus (SLE) is a typical systemic auto- of self-antigens. To investigate the role of the 5-hmC DNA immune disease, involving diffuse connective tissues (1) and modification with regard to the onset of SLE, we compared is characterized by immune inflammation. SLE has a complex the levels 5-hmC between SLE patients and normal controls. pathogenesis (2), involving genetic, immunologic and envi- Whole blood was obtained from patients, and genomic DNA ronmental factors. Thus, it may result in damage to multiple was extracted. Using the hMeDIP-chip analysis and valida- tissues and organs, especially the kidneys (3). SLE arises from tion by quantitative RT-PCR (RT-qPCR), we identified the a combination of heritable and environmental influences. differentially hydroxymethylated regions that are associated Epigenetics, the study of changes in gene expression with SLE. -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

Membrane Tension Buffering by Caveolae: a Role in Cancer?
Cancer and Metastasis Reviews (2020) 39:505–517 https://doi.org/10.1007/s10555-020-09899-2 Membrane tension buffering by caveolae: a role in cancer? Vibha Singh1 & Christophe Lamaze1 Published online: 30 May 2020 # Springer Science+Business Media, LLC, part of Springer Nature 2020 Abstract Caveolae are bulb-like invaginations made up of two essential structural proteins, caveolin-1 and cavins, which are abundantly present at the plasma membrane of vertebrate cells. Since their discovery more than 60 years ago, the function of caveolae has been mired in controversy. The last decade has seen the characterization of new caveolae components and regulators together with the discovery of additional cellular functions that have shed new light on these enigmatic structures. Early on, caveolae and/ or caveolin-1 have been involved in the regulation of several parameters associated with cancer progression such as cell migration, metastasis, angiogenesis, or cell growth. These studies have revealed that caveolin-1 and more recently cavin-1 have a dual role with either a negative or a positive effect on most of these parameters. The recent discovery that caveolae can act as mechanosensors has sparked an array of new studies that have addressed the mechanobiology of caveolae in various cellular functions. This review summarizes the current knowledge on caveolae and their role in cancer development through their activity in membrane tension buffering. We propose that the role of caveolae in cancer has to be revisited through their response to the mechanical forces encountered by cancer cells during tumor mass development. Keywords Caveolae . Cancer . Mechanosensing . Mechanotransdcution . Membrane tension . -

Mutations in EDA and EDAR Genes in a Large Mexican Hispanic Cohort with Hypohidrotic Ectodermal Dysplasia
Letter to the Editor http://dx.doi.org/10.5021/ad.2015.27.4.474 Mutations in EDA and EDAR Genes in a Large Mexican Hispanic Cohort with Hypohidrotic Ectodermal Dysplasia Julio C Salas-Alanis1,2, Eva Wozniak3, Charles A Mein3, Carola C Duran Mckinster4, Jorge Ocampo-Candiani1, David P Kelsell5, Rong Hua6, Maria L Garza-Rodriguez7, Keith A Choate6, Hugo A Barrera Saldaña7,8 1Dermatology Department, Hospital Universitario, Universidad Autonoma de Nuevo Leon, 2Basic Science Department, Medicine School, Universidad de Monterrey, Monterrey, NL, Mexico, 3Barts and the London Genome Centre, John Vane Science Centre, Barts and the London School of Medicine and Dentistry, University of London, London, United Kingdom, 4Instituto Nacional de Pediatria, Coyoacan, CP, Mexico, 5Centre for Cutaneous Research, Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom, 6Departments of Dermatology, Genetics, and Pathology, Yale University School of Medicine, New Haven, CT, USA, 7Facultad de Medicina, Departamento de Bioquímica y Medicina Molecular, Universidad Autonoma de Nuevo Leon,8Vitagenesis, Monterrey, NL, Mexico Dear Editor: cludes dryness of the skin, eyes, airways, and mucous Ectodermal dysplasias (ED) encompass nearly 200 differ- membranes, as well as other ectodermal defects and, in ent genetic conditions identified by the lack, or dysgenesis, some cases, fever, seizures, and rarely, death. of at least two ectodermal derivatives, such as hair, nails, XL-HED is caused by mutations in the EDA gene, located teeth, and sweat glands. Hypohidrotic/anhidrotic ED (HED) on chromosome Xq12-q13.1, which encodes a signaling is the most frequent form of ED and it can be inherited as molecule of the tumor necrosis factor (TNF) superfamily. -

Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins
International Journal of Molecular Sciences Article Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins M. Quadir Siddiqui 1,† , Maulik D. Badmalia 1,† and Trushar R. Patel 1,2,3,* 1 Alberta RNA Research and Training Institute, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB T1K 3M4, Canada; [email protected] (M.Q.S.); [email protected] (M.D.B.) 2 Department of Microbiology, Immunology and Infectious Disease, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada 3 Li Ka Shing Institute of Virology, University of Alberta, Edmonton, AB T6G 2E1, Canada * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Members of the human Zyxin family are LIM domain-containing proteins that perform critical cellular functions and are indispensable for cellular integrity. Despite their importance, not much is known about their structure, functions, interactions and dynamics. To provide insights into these, we used a set of in-silico tools and databases and analyzed their amino acid sequence, phylogeny, post-translational modifications, structure-dynamics, molecular interactions, and func- tions. Our analysis revealed that zyxin members are ohnologs. Presence of a conserved nuclear export signal composed of LxxLxL/LxxxLxL consensus sequence, as well as a possible nuclear localization signal, suggesting that Zyxin family members may have nuclear and cytoplasmic roles. The molecular modeling and structural analysis indicated that Zyxin family LIM domains share Citation: Siddiqui, M.Q.; Badmalia, similarities with transcriptional regulators and have positively charged electrostatic patches, which M.D.; Patel, T.R. -

A 31-Bp Indel in the 5' UTR Region of the GNB1L Gene Is Signi Cantly
A 31-bp Indel in the 5' UTR region of the GNB1L gene is signicantly associated with chicken body weight and carcass traits Tuanhui Ren South China Agricultural University Ying Yang South China Agricultural University Wujian Lin South China Agricultural University Wangyu Li South China Agricultral University Mingjian Xian South China Agricultural University Rong Fu South China Agricultural University Zihao Zhang South China Agricultural University Guodong Mo South China Agricultral University Wen Luo South China Agricultural University Xiquan Zhang ( [email protected] ) https://orcid.org/0000-0002-7940-1303 Research article Keywords: chicken, GNB1L gene, Indel, growth traits, carcass traits Posted Date: June 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-26694/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/17 Version of Record: A version of this preprint was published on August 26th, 2020. See the published version at https://doi.org/10.1186/s12863-020-00900-z. Page 2/17 Abstract Background: G-protein subunit beta 1 like (GNB1L) can encode a G-protein beta-subunit-like polypeptide, the chicken GNB1L gene is up-regulated in the breast muscle of high-feed eciency chickens, and its expression is 1.52-fold that of low-feed eciency chickens. However, there are no reports describing the effects of GNB1L gene Indel on the chicken carcass and growth traits. Results: This study identied a 31-bp Indel in 5' UTR of the GNB1L gene and elucidated the effect of this gene mutation on the carcass and growth traits in chickens. -

EHD2 Is a Mechanotransducer Connecting Caveolae Dynamics with Gene Transcription
EHD2 is a mechanotransducer connecting caveolae dynamics with gene transcription Satish Kailasam Mani, Cesar Valades-Cruz, Stéphanie Torrino, Wei-Wei Shen, Cedric Blouin, Satish Kailasam Mani, Christine Viaris de Lesegno, Pierre Bost, Alexandre Grassart, Darius Köster, et al. To cite this version: Satish Kailasam Mani, Cesar Valades-Cruz, Stéphanie Torrino, Wei-Wei Shen, Cedric Blouin, et al.. EHD2 is a mechanotransducer connecting caveolae dynamics with gene transcription. Journal of Cell Biology, Rockefeller University Press, 2018, 217 (12), pp.4092-4105. 10.1083/jcb.201801122. inserm- 02426440 HAL Id: inserm-02426440 https://www.hal.inserm.fr/inserm-02426440 Submitted on 2 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - ShareAlike| 4.0 International License REPORT EHD2 is a mechanotransducer connecting caveolae dynamics with gene transcription Stéphanie Torrino1,2,3*, Wei‑Wei Shen1,2,3*, Cédric M. Blouin1,2,3, Satish Kailasam Mani1,2,3, Christine Viaris de Lesegno1,2,3, Pierre Bost4,5, Alexandre Grassart6, Darius Köster7, Cesar Augusto Valades‑Cruz2,3,8, Valérie Chambon2,3,8, Ludger Johannes2,3,8, Paolo Pierobon9, Vassili Soumelis4, Catherine Coirault10, Stéphane Vassilopoulos10, and Christophe Lamaze1,2,3 Caveolae are small invaginated pits that function as dynamic mechanosensors to buffer tension variations at the plasma membrane. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A PAX5-OCT4-PRDM1 Developmental Switch Specifies Human Primordial Germ Cells
A PAX5-OCT4-PRDM1 Developmental Switch Specifies Human Primordial Germ Cells Fang Fang1,2, Benjamin Angulo1,2, Ninuo Xia1,2, Meena Sukhwani3, Zhengyuan Wang4, Charles C Carey5, Aurélien Mazurie5, Jun Cui1,2, Royce Wilkinson5, Blake Wiedenheft5, Naoko Irie6, M. Azim Surani6, Kyle E Orwig3, Renee A Reijo Pera1,2 1Department of Cell Biology and Neurosciences, Montana State University, Bozeman, MT 59717, USA 2Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA 3Department of Obstetrics, Gynecology and Reproductive Sciences, University of Pittsburgh, School of Medicine; Magee Women’s Research Institute, Pittsburgh, PA, 15213, USA 4Genomic Medicine Division, Hematology Branch, NHLBI/NIH, MD 20850, USA 5Department of Microbiology and Immunology, Montana State University, Bozeman, MT 59717, USA. 6Wellcome Trust Cancer Research UK Gurdon Institute, Tennis Court Road, University of Cambridge, Cambridge CB2 1QN, UK. Correspondence should be addressed to F.F. (e-mail: [email protected]) 1 Abstract Dysregulation of genetic pathways during human germ cell development leads to infertility. Here, we analyzed bona fide human primordial germ cells (hPGCs) to probe the developmental genetics of human germ cell specification and differentiation. We examined distribution of OCT4 occupancy in hPGCs relative to human embryonic stem cells (hESCs). We demonstrate that development, from pluripotent stem cells to germ cells, is driven by switching partners with OCT4 from SOX2 to PAX5 and PRDM1. Gain- and loss-of-function studies revealed that PAX5 encodes a critical regulator of hPGC development. Moreover, analysis of epistasis indicates that PAX5 acts upstream of OCT4 and PRDM1. The PAX5-OCT4-PRDM1 proteins form a core transcriptional network that activates germline and represses somatic programs during human germ cell differentiation. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -
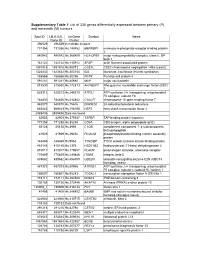
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Natural Genetic Variation Screen in Drosophila Identifies
INVESTIGATION Natural Genetic Variation Screen in Drosophila Identifies Wnt Signaling, Mitochondrial Metabolism, and Redox Homeostasis Genes as Modifiers of Apoptosis Rebecca A. S. Palu,*,1 Elaine Ong,* Kaitlyn Stevens,* Shani Chung,* Katie G. Owings,* Alan G. Goodman,†,‡ and Clement Y. Chow*,2 *Department of Human Genetics, University of Utah School of Medicine, Salt Lake City, UT 84112, †School of Molecular Biosciences, and ‡Paul G. Allen School for Global Animal Health, Washington State University College of Veterinary Medicine, Pullman, WA 99164 ORCID IDs: 0000-0001-9444-8815 (R.A.S.P.); 0000-0001-6394-332X (A.G.G.); 0000-0002-3104-7923 (C.Y.C.) ABSTRACT Apoptosis is the primary cause of degeneration in a number of neuronal, muscular, and KEYWORDS metabolic disorders. These diseases are subject to a great deal of phenotypic heterogeneity in patient apoptosis populations, primarily due to differences in genetic variation between individuals. This creates a barrier to Drosophila effective diagnosis and treatment. Understanding how genetic variation influences apoptosis could lead to genetic variation the development of new therapeutics and better personalized treatment approaches. In this study, we modifier genes examine the impact of the natural genetic variation in the Drosophila Genetic Reference Panel (DGRP) on two models of apoptosis-induced retinal degeneration: overexpression of p53 or reaper (rpr). We identify a number of known apoptotic, neural, and developmental genes as candidate modifiers of degeneration. We also use Gene Set Enrichment Analysis (GSEA) to identify pathways that harbor genetic variation that impact these apoptosis models, including Wnt signaling, mitochondrial metabolism, and redox homeostasis. Fi- nally, we demonstrate that many of these candidates have a functional effect on apoptosis and degener- ation.