Transcriptome Sequencing of Crucihimalaya Himalaica
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The First Chloroplast Genome Sequence of Boswellia Sacra, a Resin-Producing Plant in Oman
RESEARCH ARTICLE The First Chloroplast Genome Sequence of Boswellia sacra, a Resin-Producing Plant in Oman Abdul Latif Khan1, Ahmed Al-Harrasi1*, Sajjad Asaf2, Chang Eon Park2, Gun-Seok Park2, Abdur Rahim Khan2, In-Jung Lee2, Ahmed Al-Rawahi1, Jae-Ho Shin2* 1 UoN Chair of Oman's Medicinal Plants & Marine Natural Products, University of Nizwa, Nizwa, Oman, 2 School of Applied Biosciences, Kyungpook National University, Daegu, Republic of Korea a1111111111 * [email protected] (AAH); [email protected] (JHS) a1111111111 a1111111111 a1111111111 Abstract a1111111111 Boswellia sacra (Burseraceae), a keystone endemic species, is famous for the production of fragrant oleo-gum resin. However, the genetic make-up especially the genomic informa- tion about chloroplast is still unknown. Here, we described for the first time the chloroplast OPEN ACCESS (cp) genome of B. sacra. The complete cp sequence revealed a circular genome of 160,543 Citation: Khan AL, Al-Harrasi A, Asaf S, Park CE, bp size with 37.61% GC content. The cp genome is a typical quadripartite chloroplast struc- Park G-S, Khan AR, et al. (2017) The First ture with inverted repeats (IRs 26,763 bp) separated by small single copy (SSC; 18,962 bp) Chloroplast Genome Sequence of Boswellia sacra, and large single copy (LSC; 88,055 bp) regions. De novo assembly and annotation showed a Resin-Producing Plant in Oman. PLoS ONE 12 the presence of 114 unique genes with 83 protein-coding regions. The phylogenetic analysis (1): e0169794. doi:10.1371/journal.pone.0169794 revealed that the B. sacra cp genome is closely related to the cp genome of Azadirachta Editor: Xiu-Qing Li, Agriculture and Agri-Food indica and Citrus sinensis, while most of the syntenic differences were found in the non-cod- Canada, CANADA ing regions. -

Phylogenetic Position and Generic Limits of Arabidopsis (Brassicaceae)
PHYLOGENETIC POSITION Steve L. O'Kane, Jr.2 and Ihsan A. 3 AND GENERIC LIMITS OF Al-Shehbaz ARABIDOPSIS (BRASSICACEAE) BASED ON SEQUENCES OF NUCLEAR RIBOSOMAL DNA1 ABSTRACT The primary goals of this study were to assess the generic limits and monophyly of Arabidopsis and to investigate its relationships to related taxa in the family Brassicaceae. Sequences of the internal transcribed spacer region (ITS-1 and ITS-2) of nuclear ribosomal DNA, including 5.8S rDNA, were used in maximum parsimony analyses to construct phylogenetic trees. An attempt was made to include all species currently or recently included in Arabidopsis, as well as species suggested to be close relatives. Our ®ndings show that Arabidopsis, as traditionally recognized, is polyphyletic. The genus, as recircumscribed based on our results, (1) now includes species previously placed in Cardaminopsis and Hylandra as well as three species of Arabis and (2) excludes species now placed in Crucihimalaya, Beringia, Olimar- abidopsis, Pseudoarabidopsis, and Ianhedgea. Key words: Arabidopsis, Arabis, Beringia, Brassicaceae, Crucihimalaya, ITS phylogeny, Olimarabidopsis, Pseudoar- abidopsis. Arabidopsis thaliana (L.) Heynh. was ®rst rec- netic studies and has played a major role in un- ommended as a model plant for experimental ge- derstanding the various biological processes in netics over a half century ago (Laibach, 1943). In higher plants (see references in Somerville & Mey- recent years, many biologists worldwide have fo- erowitz, 2002). The intraspeci®c phylogeny of A. cused their research on this plant. As indicated by thaliana has been examined by Vander Zwan et al. Patrusky (1991), the widespread acceptance of A. (2000). Despite the acceptance of A. -

PYK10 Myrosinase Reveals a Functional Coordination Between Endoplasmic Reticulum Bodies and Glucosinolates in Arabidopsis Thaliana
The Plant Journal (2017) 89, 204–220 doi: 10.1111/tpj.13377 PYK10 myrosinase reveals a functional coordination between endoplasmic reticulum bodies and glucosinolates in Arabidopsis thaliana Ryohei T. Nakano1,2,3, Mariola Pislewska-Bednarek 4, Kenji Yamada5,†, Patrick P. Edger6,‡, Mado Miyahara3,§, Maki Kondo5, Christoph Bottcher€ 7,¶, Masashi Mori8, Mikio Nishimura5, Paul Schulze-Lefert1,2,*, Ikuko Hara-Nishimura3,*,#,k and Paweł Bednarek4,*,# 1Department of Plant Microbe Interactions, Max Planck Institute for Plant Breeding Research, Carl-von-Linne-Weg 10, D-50829 Koln,€ Germany, 2Cluster of Excellence on Plant Sciences (CEPLAS), Max Planck Institute for Plant Breeding Research, Carl-von-Linne-Weg 10, D-50829 Koln,€ Germany, 3Department of Botany, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan, 4Institute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland, 5Department of Cell Biology, National Institute of Basic Biology, Okazaki 444-8585, Japan, 6Department of Plant and Microbial Biology, University of California, Berkeley, CA 94720, USA, 7Department of Stress and Developmental Biology, Leibniz Institute of Plant Biochemistry, D-06120 Halle (Saale), Germany, and 8Ishikawa Prefectural University, Nonoichi, Ishikawa 834-1213, Japan Received 29 March 2016; revised 30 August 2016; accepted 5 September 2016; published online 19 December 2016. *For correspondence (e-mails [email protected]; [email protected]; [email protected]). #These authors contributed equally to this work. †Present address: Malopolska Centre of Biotechnology, Jagiellonian University, 30-387 Krakow, Poland. ‡Present address: Department of Horticulture, Michigan State University, East Lansing, MI, USA. §Present address: Department of Biological Sciences, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan. -

Chromosome‐Level Reference Genome of the Soursop (Annona Muricata
Received: 26 May 2020 | Revised: 5 February 2021 | Accepted: 8 February 2021 DOI: 10.1111/1755-0998.13353 RESOURCE ARTICLE Chromosome- level reference genome of the soursop (Annona muricata): A new resource for Magnoliid research and tropical pomology Joeri S. Strijk1,2,3 | Damien D. Hinsinger2,4 | Mareike M. Roeder5,6 | Lars W. Chatrou7 | Thomas L.P. Couvreur8 | Roy H.J. Erkens9 | Hervé Sauquet10 | Michael D. Pirie11 | Daniel C. Thomas12 | Kunfang Cao13 1Institute for Biodiversity and Environmental Research, Universiti Brunei Darussalam, Jalan Tungku Link, Brunei Darussalam 2Alliance for Conservation Tree Genomics, Pha Tad Ke Botanical Garden, Luang Prabang, Laos 3Guangxi Key Laboratory of Forest Ecology and Conservation, Biodiversity Genomics Team, Nanning, Guangxi, China 4Génomique Métabolique, Genoscope, Institut de Biologie François Jacob, Commissariat à l′Énergie Atomique (CEA), CNRS, Université Évry, Université Paris- Saclay, Évry, France 5Community Ecology and Conservation Group, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Menglun, Mengla, Yunnan, China 6Aueninstitut, Institute for Geography and Geoecology, Karlsruhe Institute of Technology, Rastatt, Germany 7Systematic and Evolutionary Botany Laboratory, Ghent University, Ghent, Belgium 8IRD, DIADE, Univ Montpellier, Montpellier, France 9Maastricht Science Programme, Maastricht University, Maastricht, The Netherlands 10National Herbarium of New South Wales (NSW), Royal Botanic Gardens and Domain Trust, Sydney, NSW, Australia 11Department of Natural History, -
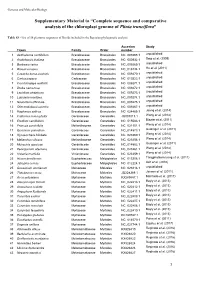
Complete Sequence and Comparative Analysis of the Chloroplast Genome of Plinia Trunciflora”
Genetics and Molecular Biology Supplementary Material to “Complete sequence and comparative analysis of the chloroplast genome of Plinia trunciflora” Table S3 - List of 56 plastome sequences of Rosids included in the Bayesian phylogenetic analysis. Accesion Study Taxon Family Order number 1 Aethionema cordifolium Brassicaceae Brassicales NC_009265.1 unpublished 2 Arabidopsis thaliana Brassicaceae Brassicales NC_000932.1 Sato et al. (1999) 3 Barbarea verna Brassicaceae Brassicales NC_009269.1 unpublished 4 Brassica napus Brassicaceae Brassicales NC_016734.1 Hu et al. (2011) 5 Capsella bursa-pastoris Brassicaceae Brassicales NC_009270.1 unpublished 6 Carica papaya Caricaceae Brassicales NC_010323.1 unpublished 7 Crucihimalaya wallichii Brassicaceae Brassicales NC_009271.1 unpublished 8 Draba nemorosa Brassicaceae Brassicales NC_009272.1 unpublished 9 Lepidium virginicum Brassicaceae Brassicales NC_009273.1 unpublished 10 Lobularia maritima Brassicaceae Brassicales NC_009274.1 unpublished 11 Nasturtium officinale Brassicaceae Brassicales NC_009275.1 unpublished 12 Olimarabidopsis pumila Brassicaceae Brassicales NC_009267.1 unpublished 13 Raphanus sativus Brassicaceae Brassicales NC_024469.1 Jeong et al. (2014) 14 California macrophylla Geraniaceae Geraniales JQ031013.1 Weng et al. (2014) 15 Erodium carvifolium Geraniaceae Geraniales NC_015083.1 Blazier et al. (2011) 16 Francoa sonchifolia Melianthaceae Geraniales NC_021101.1 Weng et al. (2014) 17 Geranium palmatum Geraniaceae Geraniales NC_014573.1 Guisinger et al. (2011) 18 Hypseocharis bilobate Geraniaceae Geraniales NC_023260.1 Weng et al. (2014) 19 Melianthus villosus Melianthaceae Geraniales NC_023256.1 Weng et al. (2014) 20 Monsonia speciose Geraniaceae Geraniales NC_014582.1 Guisinger et al. (2011) 21 Pelargonium alternans Geraniaceae Geraniales NC_023261.1 Weng et al. (2014) 22 Viviania marifolia Vivianiaceae Geraniales NC_023259.1 Weng et al. (2014) 23 Hevea brasiliensis Euphorbiaceae Malpighiales NC_015308.1 Tangphatsornruang et al. -

Downloaded from Genbank on That Full Plastid Genomes Are Not Sufficient to Reject Al- February 28, 2012
Ruhfel et al. BMC Evolutionary Biology 2014, 14:23 http://www.biomedcentral.com/1471-2148/14/23 RESEARCH ARTICLE Open Access From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes Brad R Ruhfel1*, Matthew A Gitzendanner2,3,4, Pamela S Soltis3,4, Douglas E Soltis2,3,4 and J Gordon Burleigh2,4 Abstract Background: Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae). Although these data have helped resolve the phylogeny of numerous clades (e.g., green algae, angiosperms, and gymnosperms), their utility for inferring relationships across all green plants is uncertain. Viridiplantae originated 700-1500 million years ago and may comprise as many as 500,000 species. This clade represents a major source of photosynthetic carbon and contains an immense diversity of life forms, including some of the smallest and largest eukaryotes. Here we explore the limits and challenges of inferring a comprehensive green plant phylogeny from available complete or nearly complete plastid genome sequence data. Results: We assembled protein-coding sequence data for 78 genes from 360 diverse green plant taxa with complete or nearly complete plastid genome sequences available from GenBank. Phylogenetic analyses of the plastid data recovered well-supported backbone relationships and strong support for relationships that were not observed in previous analyses of major subclades within Viridiplantae. However, there also is evidence of systematic error in some analyses. In several instances we obtained strongly supported but conflicting topologies from analyses of nucleotides versus amino acid characters, and the considerable variation in GC content among lineages and within single genomes affected the phylogenetic placement of several taxa. -

Article Resolution of Brassicaceae Phylogeny Using Nuclear Genes
Resolution of Brassicaceae Phylogeny Using Nuclear Genes Uncovers Nested Radiations and Supports Convergent Morphological Evolution Chien-Hsun Huang,1 Renran Sun,1 Yi Hu,2 Liping Zeng,1 Ning Zhang,3 Liming Cai,1 Qiang Zhang,4 Marcus A. Koch,5 Ihsan Al-Shehbaz,6 Patrick P. Edger,7 J. Chris Pires,8 Dun-Yan Tan,9 Yang Zhong,1 and Hong Ma*,1 1State Key Laboratory of Genetic Engineering and Collaborative Innovation Center of Genetics and Development, Ministry of Education Key Laboratory of Biodiversity Sciences and Ecological Engineering, Institute of Plant Biology, Institute of Biodiversity Sciences, Center for Evolutionary Biology, School of Life Sciences, Fudan University, Shanghai, China 2Department of Biology, The Huck Institute of the Life Sciences, Pennsylvania State University 3Department of Botany, National Museum of Natural History, MRC 166, Smithsonian Institution, Washington, DC 4Guangxi Key Laboratory of Plant Conservation and Restoration Ecology in Karst Terrain, Guangxi Institute of Botany, Guangxi Zhuang Autonomous Region and Chinese Academy of Sciences, Guilin, China 5Biodiversity and Plant Systematics, Centre for Organismal Studies Heidelberg, Heidelberg University, Heidelberg, Germany 6Missouri Botanical Garden, St. Louis 7Department of Horticulture, Michigan State University, East Lansing 8Division of Biological Sciences, University of Missouri, Columbia 9Xinjiang Key Laboratory of Grassland Resources and Ecology, College of Grassland and Environment Sciences, Xinjiang Agricultural University, Ur€ umqi,€ China *Corresponding author: E-mail: [email protected]. Associate editor: Hongzhi Kong Abstract Brassicaceae is one of the most diverse and economically valuable angiosperm families with widely cultivated vegetable crops and scientifically important model plants, such as Arabidopsis thaliana. The evolutionary history, ecological, morphological, and genetic diversity, and abundant resources and knowledge of Brassicaceae make it an excellent model family for evolutionary studies. -

Conspectus of the Vascular Plants of Mongolia” (2014)
Turczaninowia 18 (2): 39–67 (2015) ISSN 1560–7259 (print edition) DOI: 10.14258/turczaninowia.18.2.4 TURCZANINOWIA www.ssbg.asu.ru/turczaninowia.php ISSN 1560–7267 (online edition) УДК 581.9 (582.683.2/517.3) Cruciferae (Brassicaceae): Alternative treatment for the “Conspectus of the vascular plants of Mongolia” (2014) Cruciferae (Brassicaceae): альтернативная обработка для «Конспекта сосудистых растений Монголии» (2014) D.A. German Д.А. Герман Центр исследований организмов, Гейдельбергский университет, Им Нойенхаймер Фельд, 345; D-69120, Гейдельберг, Германия; Южно-Сибирский ботанический сад, Алтайский государственный университет, пр-т Ленина, 61; 656049, Барнаул, Россия. E-mail: [email protected] Centre for Organismal Studies, Heidelberg University, Im Neuenheimer Feld 345; D-69120 Heidelberg, Germany; South-Siberian Botanical Garden, Altai State University, Lenina str., 61; 656049, Barnaul, Russia Key words: Central Asia, phytodiversity, floristic findings, new combinations, systematics, Guenthera, Matthiola, Stevenia. Ключевые слова: Центральная Азия, фиторазнообразие, флористические находки, новые комбинации, систе- матика, Guenthera, Matthiola, Stevenia. Summary. The checklist of the mustards (Cruciferae, или отсутствию в каждом из 16-ти ботанико-геогра- or Brassicaceae) of Mongolia supplied with the data on фических регионов) в Монголии. Впервые для стра- their distribution according to the phytogeographical re- ны указан род левкой – Matthiola W. T. Aiton, пред- gions of the country is presented which includes 141 spe- ставленный естественно произрастающим здесь M. cies and 7 subspecies from 59 genera. Both taxonomic superba Conti; некоторые виды отмечены в качестве diversity and geography of Mongolian Cruciferae are новинок для отдельных районов. В то же время, пере- critically revised with use of herbarium material, online числены и прокомментированы ошибочные указания resources, and published data. -

Publications of Ihsan A
TAXA NAMED IN HONOR OF IHSAN A. AL-SHEHBAZ 1. Tribe Shehbazieae D. A. German, Turczaninowia 17(4): 22. 2014. 2. Shehbazia D. A. German, Turczaninowia 17(4): 20. 2014. 3. Shehbazia tibetica (Maxim.) D. A. German, Turczaninowia 17(4): 20. 2014. 4. Astragalus shehbazii Zarre & Podlech, Feddes Repert. 116: 70. 2005. 5. Bornmuellerantha alshehbaziana Dönmez & Mutlu, Novon 20: 265. 2010. 6. Centaurea shahbazii Ranjbar & Negaresh, Edinb. J. Bot. 71: 1. 2014. 7. Draba alshehbazii Klimeš & D. A. German, Bot. J. Linn. Soc. 158: 750. 2008. 8. Euphorbia shehbaziana S.Y.Hama & S.A.Ahmad, Harvard Pap. Bot. 25: 73. 2020. 9. Ferula shehbaziana S. A. Ahmad, Harvard Pap. Bot. 18: 99. 2013. 10. Matthiola shehbazii Ranjbar & Karami, Nordic J. Bot. 32: 714. 2013. 11. Plocama alshehbazii F. O. Khass., D. Khamr., U. Khuzh. & Achilova, Stapfia 101: 25. 2014. 12. Alshehbazia Salariato & Zuloaga, Kew Bulletin 70: 5. 2015 13. Alshehbzia hauthalii (Gilg & Muschl.) Salariato & Zuloaga, Kew Bulletin 70: 5. 2015 14. Ihsanalshehbazia Tahir Ali & Thines, Taxon 65: 93. 2016. 15. Ihsanalshehbazia granatensis (Boiss. & Reuter) Tahir Ali & Thines, Taxon 65. 93. 2016. 16. Aubrieta alshehbazii Dönmez, Ugurlu & M.A.Koch, Phytotaxa 299. 104. 2017. 17. Silene shehbazii S.A.Ahmad, Novon 25: 131. 2017. PUBLICATIONS OF IHSAN A. AL-SHEHBAZ 1973 1. Al-Shehbaz, I. A. 1973. The biosystematics of the genus Thelypodium (Cruciferae). Contrib. Gray Herb. 204: 3-148. 1977 2. Al-Shehbaz, I. A. 1977. Protogyny, Cruciferae. Syst. Bot. 2: 327-333. 3. A. R. Al-Mayah & I. A. Al-Shehbaz. 1977. Chromosome numbers for some Leguminosae from Iraq. -

Transcriptomes of Saussurea (Asteraceae) Provide Insights Into High-Altitude Adaptation
plants Article Transcriptomes of Saussurea (Asteraceae) Provide Insights into High-Altitude Adaptation Xu Zhang 1,2,3 , Yanxia Sun 1,2, Jacob B. Landis 4,5 , Jun Shen 6, Huajie Zhang 1,2, Tianhui Kuang 7, Wenguang Sun 7,8 , Jiao Sun 1,2, Bashir B. Tiamiyu 1,2 , Tao Deng 7, Hang Sun 7,* and Hengchang Wang 1,2,* 1 CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; [email protected] (X.Z.); [email protected] (Y.S.); [email protected] (H.Z.); [email protected] (J.S.); [email protected] (B.B.T.) 2 Center of Conservation Biology, Core Botanical Gardens, Chinese Academy of Sciences, Wuhan 430074, China 3 University of Chinese Academy of Sciences, Beijing 100049, China 4 Section of Plant Biology and the L.H. Bailey Hortorium, School of Integrative Plant Science, Cornell University, Ithaca, NY 14850, USA; [email protected] 5 BTI Computational Biology Center, Boyce Thompson Institute, Ithaca, NY 14853, USA 6 State Key Laboratory of Rice Biology, Zhejiang Provincial Key Laboratory of Crop Genetic Resources, Institute of Crop Science, College of Agriculture and Biotechnology, Zhejiang University, Hangzhou 310058, China; [email protected] 7 Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China; [email protected] (T.K.); [email protected] (W.S.); [email protected] (T.D.) 8 School of Life Science, Yunnan Normal University, Yunnan 650500, China * Correspondence: [email protected] (H.S.); [email protected] (H.W.) Citation: Zhang, X.; Sun, Y.; Landis, J.B.; Shen, J.; Zhang, H.; Kuang, T.; Abstract: Understanding how species adapt to extreme environments is an extension of the main Sun, W.; Sun, J.; Tiamiyu, B.B.; Deng, goals of evolutionary biology. -

Mandakova TPC2010, Suppl Mat.Pdf
Supplemental Data. Mandáková et al. (2010). Plant Cell 10.1105/tpc.110.074526 Supplemental Figure 1. A Three-Way Comparison of the Relative Position of Corresponding Synteny Blocks of Stenopetalum nutans (SN), S. lineare (SL) and Ballantinia antipoda (BA) Relative to the Reference Ancestral Crucifer Karyotype (ACK). In the main panel of the image, each of the three modern karyotypes is presented in a radial layout. Within each of the three karyotypes, ideograms are ordered and oriented in the outward direction (corresponding to the same counter-clockwise scale progression of Figure 3). Each line connects a pair of genomic positions on two different modern genomes that are syntenically related to the same genomic block in the ACK. For example, the light-blue line at the top of the figure between SL and SN corresponds to synteny with the genomic block U2 on AK7. Each of the eight small panels shows synteny relationships between the three modern genomes for a specific ancestral chromosome (AK1-8). Supplemental Data. Mandáková et al. (2010). Plant Cell 10.1105/tpc.110.074526 Supplemental Figure 2. The Unique Rearrangement of the AK8(#1)-like Homoeologue Shared by All Analyzed Species. This rearrangement was mediated by two subsequent paracentric inversions involving two thirds of genomic block W1 and a major part of X1. In S. lineare, block V1 underwent a secondary translocation to another chromosome. The rearrangement not shown for Arabidella eremigena and Blennodia canescens. Supplemental Data. Mandáková et al. (2010). Plant Cell 10.1105/tpc.110.074526 Supplemental Figure 3. Phylogeny of the Malate Synthase (MS) (TrN + Γ + I) Showing the Position of Sequences from the Australian Species (in Bold) in the Context of Other Brassicaceae Taxa. -

Brassicaceae
Wild Crucifer Species as Sources of Traits Wild Crucifer Species as Sources of Agronomic Traits The following guide to the wild germplasm of Brassica and allied crops reviews the potential of wild crucifers, particularly members of the tribe Brassiceae, as sources of agronomic traits. In addition to traditional breeding methods, interspecific and intergeneric transfer of genes governing qualitative and quantitative characters from wild allies to cultivated forms will be facilitated with various in vitro methods, such as somatic cell genetics and recombinant DNA techniques (See Guide Part III). Examples of genetic variability in potential agronomic traits of germplasms of Brassica and related genera (Tribe Brassiceae) will be presented under 14 sections. Data will be presented for all species in the Family. CONTENTS 1. GERMPLASM 1.1 Brassicaceae Species Checklist 1.2 North American Germplasm – Tribe Brassiceae 1.2.1 North American Ethnobotany 1.2.2 Weed and Crop Species in Canada 1.2.3 Weed and Crop Species in U.S.A. and Mexico 1.2.4 Invasives 2. MORPHOLOGICAL CHARACTERS 2.1 Hairs/Trichomes 2.2 Leaf Thickness/Waxiness 2.3 Petal Colour 2.4 Resistance to Pod Shattering 2.5 Growth Form and Geocarpy 2.6 Floral Structure 2.7 Fruit Type and Dispersal 2.8 Seed Size 2.9 Vegetative Reproduction 3. GENOME ARRANGEMENT 3.1 Chromosome Numbers 3.2 Genome Size 3.3 Genome Comparative Mapping 3.4 Cruciferae: Compendium of Trait Genetics 4. CHEMICAL TRAITS 4.1 Fatty Acids 4.2 Glucosinolates (mustard oil glucosides) 4.3 Phenolics 4.4 Secondary Metabolites/Floral Pigments 4.5 Anti-pest/Allelopathy 4.6 Flavonoids 4.7 Peroxidases 4.8 Tocopherols 4.9 Nectar Production Wild Crucifer Species as Sources of Traits 4.10 Mucilage 4.11 Cyanide 5.