Transcriptomes of Saussurea (Asteraceae) Provide Insights Into High-Altitude Adaptation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The First Chloroplast Genome Sequence of Boswellia Sacra, a Resin-Producing Plant in Oman
RESEARCH ARTICLE The First Chloroplast Genome Sequence of Boswellia sacra, a Resin-Producing Plant in Oman Abdul Latif Khan1, Ahmed Al-Harrasi1*, Sajjad Asaf2, Chang Eon Park2, Gun-Seok Park2, Abdur Rahim Khan2, In-Jung Lee2, Ahmed Al-Rawahi1, Jae-Ho Shin2* 1 UoN Chair of Oman's Medicinal Plants & Marine Natural Products, University of Nizwa, Nizwa, Oman, 2 School of Applied Biosciences, Kyungpook National University, Daegu, Republic of Korea a1111111111 * [email protected] (AAH); [email protected] (JHS) a1111111111 a1111111111 a1111111111 Abstract a1111111111 Boswellia sacra (Burseraceae), a keystone endemic species, is famous for the production of fragrant oleo-gum resin. However, the genetic make-up especially the genomic informa- tion about chloroplast is still unknown. Here, we described for the first time the chloroplast OPEN ACCESS (cp) genome of B. sacra. The complete cp sequence revealed a circular genome of 160,543 Citation: Khan AL, Al-Harrasi A, Asaf S, Park CE, bp size with 37.61% GC content. The cp genome is a typical quadripartite chloroplast struc- Park G-S, Khan AR, et al. (2017) The First ture with inverted repeats (IRs 26,763 bp) separated by small single copy (SSC; 18,962 bp) Chloroplast Genome Sequence of Boswellia sacra, and large single copy (LSC; 88,055 bp) regions. De novo assembly and annotation showed a Resin-Producing Plant in Oman. PLoS ONE 12 the presence of 114 unique genes with 83 protein-coding regions. The phylogenetic analysis (1): e0169794. doi:10.1371/journal.pone.0169794 revealed that the B. sacra cp genome is closely related to the cp genome of Azadirachta Editor: Xiu-Qing Li, Agriculture and Agri-Food indica and Citrus sinensis, while most of the syntenic differences were found in the non-cod- Canada, CANADA ing regions. -

Phylogenetic Position and Generic Limits of Arabidopsis (Brassicaceae)
PHYLOGENETIC POSITION Steve L. O'Kane, Jr.2 and Ihsan A. 3 AND GENERIC LIMITS OF Al-Shehbaz ARABIDOPSIS (BRASSICACEAE) BASED ON SEQUENCES OF NUCLEAR RIBOSOMAL DNA1 ABSTRACT The primary goals of this study were to assess the generic limits and monophyly of Arabidopsis and to investigate its relationships to related taxa in the family Brassicaceae. Sequences of the internal transcribed spacer region (ITS-1 and ITS-2) of nuclear ribosomal DNA, including 5.8S rDNA, were used in maximum parsimony analyses to construct phylogenetic trees. An attempt was made to include all species currently or recently included in Arabidopsis, as well as species suggested to be close relatives. Our ®ndings show that Arabidopsis, as traditionally recognized, is polyphyletic. The genus, as recircumscribed based on our results, (1) now includes species previously placed in Cardaminopsis and Hylandra as well as three species of Arabis and (2) excludes species now placed in Crucihimalaya, Beringia, Olimar- abidopsis, Pseudoarabidopsis, and Ianhedgea. Key words: Arabidopsis, Arabis, Beringia, Brassicaceae, Crucihimalaya, ITS phylogeny, Olimarabidopsis, Pseudoar- abidopsis. Arabidopsis thaliana (L.) Heynh. was ®rst rec- netic studies and has played a major role in un- ommended as a model plant for experimental ge- derstanding the various biological processes in netics over a half century ago (Laibach, 1943). In higher plants (see references in Somerville & Mey- recent years, many biologists worldwide have fo- erowitz, 2002). The intraspeci®c phylogeny of A. cused their research on this plant. As indicated by thaliana has been examined by Vander Zwan et al. Patrusky (1991), the widespread acceptance of A. (2000). Despite the acceptance of A. -

PYK10 Myrosinase Reveals a Functional Coordination Between Endoplasmic Reticulum Bodies and Glucosinolates in Arabidopsis Thaliana
The Plant Journal (2017) 89, 204–220 doi: 10.1111/tpj.13377 PYK10 myrosinase reveals a functional coordination between endoplasmic reticulum bodies and glucosinolates in Arabidopsis thaliana Ryohei T. Nakano1,2,3, Mariola Pislewska-Bednarek 4, Kenji Yamada5,†, Patrick P. Edger6,‡, Mado Miyahara3,§, Maki Kondo5, Christoph Bottcher€ 7,¶, Masashi Mori8, Mikio Nishimura5, Paul Schulze-Lefert1,2,*, Ikuko Hara-Nishimura3,*,#,k and Paweł Bednarek4,*,# 1Department of Plant Microbe Interactions, Max Planck Institute for Plant Breeding Research, Carl-von-Linne-Weg 10, D-50829 Koln,€ Germany, 2Cluster of Excellence on Plant Sciences (CEPLAS), Max Planck Institute for Plant Breeding Research, Carl-von-Linne-Weg 10, D-50829 Koln,€ Germany, 3Department of Botany, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan, 4Institute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland, 5Department of Cell Biology, National Institute of Basic Biology, Okazaki 444-8585, Japan, 6Department of Plant and Microbial Biology, University of California, Berkeley, CA 94720, USA, 7Department of Stress and Developmental Biology, Leibniz Institute of Plant Biochemistry, D-06120 Halle (Saale), Germany, and 8Ishikawa Prefectural University, Nonoichi, Ishikawa 834-1213, Japan Received 29 March 2016; revised 30 August 2016; accepted 5 September 2016; published online 19 December 2016. *For correspondence (e-mails [email protected]; [email protected]; [email protected]). #These authors contributed equally to this work. †Present address: Malopolska Centre of Biotechnology, Jagiellonian University, 30-387 Krakow, Poland. ‡Present address: Department of Horticulture, Michigan State University, East Lansing, MI, USA. §Present address: Department of Biological Sciences, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan. -

Chromosome‐Level Reference Genome of the Soursop (Annona Muricata
Received: 26 May 2020 | Revised: 5 February 2021 | Accepted: 8 February 2021 DOI: 10.1111/1755-0998.13353 RESOURCE ARTICLE Chromosome- level reference genome of the soursop (Annona muricata): A new resource for Magnoliid research and tropical pomology Joeri S. Strijk1,2,3 | Damien D. Hinsinger2,4 | Mareike M. Roeder5,6 | Lars W. Chatrou7 | Thomas L.P. Couvreur8 | Roy H.J. Erkens9 | Hervé Sauquet10 | Michael D. Pirie11 | Daniel C. Thomas12 | Kunfang Cao13 1Institute for Biodiversity and Environmental Research, Universiti Brunei Darussalam, Jalan Tungku Link, Brunei Darussalam 2Alliance for Conservation Tree Genomics, Pha Tad Ke Botanical Garden, Luang Prabang, Laos 3Guangxi Key Laboratory of Forest Ecology and Conservation, Biodiversity Genomics Team, Nanning, Guangxi, China 4Génomique Métabolique, Genoscope, Institut de Biologie François Jacob, Commissariat à l′Énergie Atomique (CEA), CNRS, Université Évry, Université Paris- Saclay, Évry, France 5Community Ecology and Conservation Group, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Menglun, Mengla, Yunnan, China 6Aueninstitut, Institute for Geography and Geoecology, Karlsruhe Institute of Technology, Rastatt, Germany 7Systematic and Evolutionary Botany Laboratory, Ghent University, Ghent, Belgium 8IRD, DIADE, Univ Montpellier, Montpellier, France 9Maastricht Science Programme, Maastricht University, Maastricht, The Netherlands 10National Herbarium of New South Wales (NSW), Royal Botanic Gardens and Domain Trust, Sydney, NSW, Australia 11Department of Natural History, -

Threats to Australia's Grazing Industries by Garden
final report Project Code: NBP.357 Prepared by: Jenny Barker, Rod Randall,Tony Grice Co-operative Research Centre for Australian Weed Management Date published: May 2006 ISBN: 1 74036 781 2 PUBLISHED BY Meat and Livestock Australia Limited Locked Bag 991 NORTH SYDNEY NSW 2059 Weeds of the future? Threats to Australia’s grazing industries by garden plants Meat & Livestock Australia acknowledges the matching funds provided by the Australian Government to support the research and development detailed in this publication. This publication is published by Meat & Livestock Australia Limited ABN 39 081 678 364 (MLA). Care is taken to ensure the accuracy of the information contained in this publication. However MLA cannot accept responsibility for the accuracy or completeness of the information or opinions contained in the publication. You should make your own enquiries before making decisions concerning your interests. Reproduction in whole or in part of this publication is prohibited without prior written consent of MLA. Weeds of the future? Threats to Australia’s grazing industries by garden plants Abstract This report identifies 281 introduced garden plants and 800 lower priority species that present a significant risk to Australia’s grazing industries should they naturalise. Of the 281 species: • Nearly all have been recorded overseas as agricultural or environmental weeds (or both); • More than one tenth (11%) have been recorded as noxious weeds overseas; • At least one third (33%) are toxic and may harm or even kill livestock; • Almost all have been commercially available in Australia in the last 20 years; • Over two thirds (70%) were still available from Australian nurseries in 2004; • Over two thirds (72%) are not currently recognised as weeds under either State or Commonwealth legislation. -

The Naturalized Vascular Plants of Western Australia 1
12 Plant Protection Quarterly Vol.19(1) 2004 Distribution in IBRA Regions Western Australia is divided into 26 The naturalized vascular plants of Western Australia natural regions (Figure 1) that are used for 1: Checklist, environmental weeds and distribution in bioregional planning. Weeds are unevenly distributed in these regions, generally IBRA regions those with the greatest amount of land disturbance and population have the high- Greg Keighery and Vanda Longman, Department of Conservation and Land est number of weeds (Table 4). For exam- Management, WA Wildlife Research Centre, PO Box 51, Wanneroo, Western ple in the tropical Kimberley, VB, which Australia 6946, Australia. contains the Ord irrigation area, the major cropping area, has the greatest number of weeds. However, the ‘weediest regions’ are the Swan Coastal Plain (801) and the Abstract naturalized, but are no longer considered adjacent Jarrah Forest (705) which contain There are 1233 naturalized vascular plant naturalized and those taxa recorded as the capital Perth, several other large towns taxa recorded for Western Australia, com- garden escapes. and most of the intensive horticulture of posed of 12 Ferns, 15 Gymnosperms, 345 A second paper will rank the impor- the State. Monocotyledons and 861 Dicotyledons. tance of environmental weeds in each Most of the desert has low numbers of Of these, 677 taxa (55%) are environmen- IBRA region. weeds, ranging from five recorded for the tal weeds, recorded from natural bush- Gibson Desert to 135 for the Carnarvon land areas. Another 94 taxa are listed as Results (containing the horticultural centre of semi-naturalized garden escapes. Most Total naturalized flora Carnarvon). -

On the Flora of Australia
L'IBRARY'OF THE GRAY HERBARIUM HARVARD UNIVERSITY. BOUGHT. THE FLORA OF AUSTRALIA, ITS ORIGIN, AFFINITIES, AND DISTRIBUTION; BEING AN TO THE FLORA OF TASMANIA. BY JOSEPH DALTON HOOKER, M.D., F.R.S., L.S., & G.S.; LATE BOTANIST TO THE ANTARCTIC EXPEDITION. LONDON : LOVELL REEVE, HENRIETTA STREET, COVENT GARDEN. r^/f'ORElGN&ENGLISH' <^ . 1859. i^\BOOKSELLERS^.- PR 2G 1.912 Gray Herbarium Harvard University ON THE FLORA OF AUSTRALIA ITS ORIGIN, AFFINITIES, AND DISTRIBUTION. I I / ON THE FLORA OF AUSTRALIA, ITS ORIGIN, AFFINITIES, AND DISTRIBUTION; BEIKG AN TO THE FLORA OF TASMANIA. BY JOSEPH DALTON HOOKER, M.D., F.R.S., L.S., & G.S.; LATE BOTANIST TO THE ANTARCTIC EXPEDITION. Reprinted from the JJotany of the Antarctic Expedition, Part III., Flora of Tasmania, Vol. I. LONDON : LOVELL REEVE, HENRIETTA STREET, COVENT GARDEN. 1859. PRINTED BY JOHN EDWARD TAYLOR, LITTLE QUEEN STREET, LINCOLN'S INN FIELDS. CONTENTS OF THE INTRODUCTORY ESSAY. § i. Preliminary Remarks. PAGE Sources of Information, published and unpublished, materials, collections, etc i Object of arranging them to discuss the Origin, Peculiarities, and Distribution of the Vegetation of Australia, and to regard them in relation to the views of Darwin and others, on the Creation of Species .... iii^ § 2. On the General Phenomena of Variation in the Vegetable Kingdom. All plants more or less variable ; rate, extent, and nature of variability ; differences of amount and degree in different natural groups of plants v Parallelism of features of variability in different groups of individuals (varieties, species, genera, etc.), and in wild and cultivated plants vii Variation a centrifugal force ; the tendency in the progeny of varieties being to depart further from their original types, not to revert to them viii Effects of cross-impregnation and hybridization ultimately favourable to permanence of specific character x Darwin's Theory of Natural Selection ; — its effects on variable organisms under varying conditions is to give a temporary stability to races, species, genera, etc xi § 3. -

Effect of Gundelia Tournefortii L. on Some Cardiovascular Risk Factors in an Animal Model
J Herbmed Pharmacol. 2017; 6(4): 191-195. Journal of Herbmed Pharmacology Journal homepage: http://www.herbmedpharmacol.com Effect of Gundelia tournefortii L. on some cardiovascular risk factors in an animal model Laleh Rafiee1, Mahtab Keshvari2, Ahmad Movahedian Atar3, Zahra Hamidzadeh1, Gholam Reza Dashti1, Mahmoud Rafieian-Kopaei4, Sedigheh Asgary2* 1Physiology Research Centre, Isfahan University of Medical Sciences, Isfahan, Iran 2Isfahan Cardiovascular Research Center, Cardiovascular Research Institute, Isfahan University of Medical Sciences, Isfahan, Iran 3School of pharmacy and pharmaceutical research center, Isfahan University of Medical Sciences, Isfahan, Iran 4Medical Plants Research Center, Basic Health Sciences Institute, Shahrekord University of Medical Sciences, Shahrekord, Iran A R T I C L E I N F O A B S T R A C T Article Type: Introduction: There is no certain result in the field of industrial pharmacy approaching to Original find effective drugs in prevention and treatment of atherosclerosis, like the control of lipid factors that are major risk factors of atherosclerosis. This study was designed to evaluate the Article History: effects of Gundelia tournefortii on atherosclerosis biomarkers by measuring some biochemical Received: 4 January 2017 factors and formation of fatty streaks. Accepted: 26 August 2017 Methods: In this preclinical study, 20 male rabbits were randomly divided into 4 groups as normal diet, cholesterol diet, normal diet supplemented with G. tournefortii extract, and Keywords: cholesterol diet supplemented with G. tournefortii extract. Fasting blood samples were taken Gundelia tournefortii at the first and end of the study. Lipoproteins, Apo lipoproteins, fasting blood sugar (FBS), low Atherosclerosis density lipoproteins (LDL), factor VII and C-reactive protein (CRP) were measured. -

The Tribe Cichorieae In
Chapter24 Cichorieae Norbert Kilian, Birgit Gemeinholzer and Hans Walter Lack INTRODUCTION general lines seem suffi ciently clear so far, our knowledge is still insuffi cient regarding a good number of questions at Cichorieae (also known as Lactuceae Cass. (1819) but the generic rank as well as at the evolution of the tribe. name Cichorieae Lam. & DC. (1806) has priority; Reveal 1997) are the fi rst recognized and perhaps taxonomically best studied tribe of Compositae. Their predominantly HISTORICAL OVERVIEW Holarctic distribution made the members comparatively early known to science, and the uniform character com- Tournefort (1694) was the fi rst to recognize and describe bination of milky latex and homogamous capitula with Cichorieae as a taxonomic entity, forming the thirteenth 5-dentate, ligulate fl owers, makes the members easy to class of the plant kingdom and, remarkably, did not in- identify. Consequently, from the time of initial descrip- clude a single plant now considered outside the tribe. tion (Tournefort 1694) until today, there has been no dis- This refl ects the convenient recognition of the tribe on agreement about the overall circumscription of the tribe. the basis of its homogamous ligulate fl owers and latex. He Nevertheless, the tribe in this traditional circumscription called the fl ower “fl os semifl osculosus”, paid particular at- is paraphyletic as most recent molecular phylogenies have tention to the pappus and as a consequence distinguished revealed. Its circumscription therefore is, for the fi rst two groups, the fi rst to comprise plants with a pappus, the time, changed in the present treatment. second those without. -
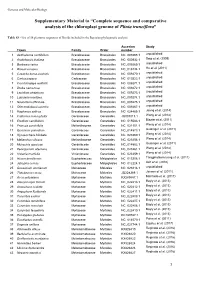
Complete Sequence and Comparative Analysis of the Chloroplast Genome of Plinia Trunciflora”
Genetics and Molecular Biology Supplementary Material to “Complete sequence and comparative analysis of the chloroplast genome of Plinia trunciflora” Table S3 - List of 56 plastome sequences of Rosids included in the Bayesian phylogenetic analysis. Accesion Study Taxon Family Order number 1 Aethionema cordifolium Brassicaceae Brassicales NC_009265.1 unpublished 2 Arabidopsis thaliana Brassicaceae Brassicales NC_000932.1 Sato et al. (1999) 3 Barbarea verna Brassicaceae Brassicales NC_009269.1 unpublished 4 Brassica napus Brassicaceae Brassicales NC_016734.1 Hu et al. (2011) 5 Capsella bursa-pastoris Brassicaceae Brassicales NC_009270.1 unpublished 6 Carica papaya Caricaceae Brassicales NC_010323.1 unpublished 7 Crucihimalaya wallichii Brassicaceae Brassicales NC_009271.1 unpublished 8 Draba nemorosa Brassicaceae Brassicales NC_009272.1 unpublished 9 Lepidium virginicum Brassicaceae Brassicales NC_009273.1 unpublished 10 Lobularia maritima Brassicaceae Brassicales NC_009274.1 unpublished 11 Nasturtium officinale Brassicaceae Brassicales NC_009275.1 unpublished 12 Olimarabidopsis pumila Brassicaceae Brassicales NC_009267.1 unpublished 13 Raphanus sativus Brassicaceae Brassicales NC_024469.1 Jeong et al. (2014) 14 California macrophylla Geraniaceae Geraniales JQ031013.1 Weng et al. (2014) 15 Erodium carvifolium Geraniaceae Geraniales NC_015083.1 Blazier et al. (2011) 16 Francoa sonchifolia Melianthaceae Geraniales NC_021101.1 Weng et al. (2014) 17 Geranium palmatum Geraniaceae Geraniales NC_014573.1 Guisinger et al. (2011) 18 Hypseocharis bilobate Geraniaceae Geraniales NC_023260.1 Weng et al. (2014) 19 Melianthus villosus Melianthaceae Geraniales NC_023256.1 Weng et al. (2014) 20 Monsonia speciose Geraniaceae Geraniales NC_014582.1 Guisinger et al. (2011) 21 Pelargonium alternans Geraniaceae Geraniales NC_023261.1 Weng et al. (2014) 22 Viviania marifolia Vivianiaceae Geraniales NC_023259.1 Weng et al. (2014) 23 Hevea brasiliensis Euphorbiaceae Malpighiales NC_015308.1 Tangphatsornruang et al. -

Downloaded from Genbank on That Full Plastid Genomes Are Not Sufficient to Reject Al- February 28, 2012
Ruhfel et al. BMC Evolutionary Biology 2014, 14:23 http://www.biomedcentral.com/1471-2148/14/23 RESEARCH ARTICLE Open Access From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes Brad R Ruhfel1*, Matthew A Gitzendanner2,3,4, Pamela S Soltis3,4, Douglas E Soltis2,3,4 and J Gordon Burleigh2,4 Abstract Background: Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae). Although these data have helped resolve the phylogeny of numerous clades (e.g., green algae, angiosperms, and gymnosperms), their utility for inferring relationships across all green plants is uncertain. Viridiplantae originated 700-1500 million years ago and may comprise as many as 500,000 species. This clade represents a major source of photosynthetic carbon and contains an immense diversity of life forms, including some of the smallest and largest eukaryotes. Here we explore the limits and challenges of inferring a comprehensive green plant phylogeny from available complete or nearly complete plastid genome sequence data. Results: We assembled protein-coding sequence data for 78 genes from 360 diverse green plant taxa with complete or nearly complete plastid genome sequences available from GenBank. Phylogenetic analyses of the plastid data recovered well-supported backbone relationships and strong support for relationships that were not observed in previous analyses of major subclades within Viridiplantae. However, there also is evidence of systematic error in some analyses. In several instances we obtained strongly supported but conflicting topologies from analyses of nucleotides versus amino acid characters, and the considerable variation in GC content among lineages and within single genomes affected the phylogenetic placement of several taxa. -

Article Resolution of Brassicaceae Phylogeny Using Nuclear Genes
Resolution of Brassicaceae Phylogeny Using Nuclear Genes Uncovers Nested Radiations and Supports Convergent Morphological Evolution Chien-Hsun Huang,1 Renran Sun,1 Yi Hu,2 Liping Zeng,1 Ning Zhang,3 Liming Cai,1 Qiang Zhang,4 Marcus A. Koch,5 Ihsan Al-Shehbaz,6 Patrick P. Edger,7 J. Chris Pires,8 Dun-Yan Tan,9 Yang Zhong,1 and Hong Ma*,1 1State Key Laboratory of Genetic Engineering and Collaborative Innovation Center of Genetics and Development, Ministry of Education Key Laboratory of Biodiversity Sciences and Ecological Engineering, Institute of Plant Biology, Institute of Biodiversity Sciences, Center for Evolutionary Biology, School of Life Sciences, Fudan University, Shanghai, China 2Department of Biology, The Huck Institute of the Life Sciences, Pennsylvania State University 3Department of Botany, National Museum of Natural History, MRC 166, Smithsonian Institution, Washington, DC 4Guangxi Key Laboratory of Plant Conservation and Restoration Ecology in Karst Terrain, Guangxi Institute of Botany, Guangxi Zhuang Autonomous Region and Chinese Academy of Sciences, Guilin, China 5Biodiversity and Plant Systematics, Centre for Organismal Studies Heidelberg, Heidelberg University, Heidelberg, Germany 6Missouri Botanical Garden, St. Louis 7Department of Horticulture, Michigan State University, East Lansing 8Division of Biological Sciences, University of Missouri, Columbia 9Xinjiang Key Laboratory of Grassland Resources and Ecology, College of Grassland and Environment Sciences, Xinjiang Agricultural University, Ur€ umqi,€ China *Corresponding author: E-mail: [email protected]. Associate editor: Hongzhi Kong Abstract Brassicaceae is one of the most diverse and economically valuable angiosperm families with widely cultivated vegetable crops and scientifically important model plants, such as Arabidopsis thaliana. The evolutionary history, ecological, morphological, and genetic diversity, and abundant resources and knowledge of Brassicaceae make it an excellent model family for evolutionary studies.