Development of Polyhipe Chromatography And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Belgian Laces
Belgian Laces “Le Gros-Chêne”, the Old Oak Tree, around 1875 – from a painting by Auguste Barbier http://ibelgique.ifrance.com/arbresdumonde/chene_set.htm Volume 17 # 65 December 1995 BELGIAN LACES ISSN 1046-0462 Official Quarterly Bulletin of THE BELGIAN RESEARCHERS Belgian American Heritage Association Founded in 1976 Our principal objective is: Keep the Belgian Heritage alive in our hearts and in the hearts of our posterity President Pierre Inghels Vice-President Micheline Gaudette Assistant VP Leen Inghels Treasurer Marlena Bellavia Secretary Patricia Robinson Dues to THE BELGIAN RESEARCHERS with subscription to BELGIAN LACES Are: In the US $12.00 a year In Canada $12.00 a year in US funds Other Countries $14.00 a year in US funds Subscribers in Europe, please add US $4.00 if you wish to receive your magazine per airmail. All subscriptions are for the calendar year. New subscribers receive the four issues of the current year, regardless when paid. Opinions expressed in Belgian Laces are not necessarily those of The Belgian Researchers or of the staff. TABLE OF CONTENTS Member portrait: Don DALEBROUX 62 A Gold Mine of Data, Georges PICAVET 63 Le Vieux Chene, Leen INGHELS 63 Sheldon, NY, Micheline GAUDETTE 65 Wisconsin Corner, Mary Ann Defnet, 70 Perfect Timing, Don VAN HOUDENOS 72 Henry VERSLYPE, Pierre INGHELS 73 Belgo-American Centenarian, Leen INGHELS 74 WWII Memories, John VAN DORPE 74 Where in Cyberspace is Belgium?, Hans Michael VERMEERSCH 75 Those Wacky Walloons!, Leen INGHELS 76 Manneken Pis 77 Passenger Lists, M. GAUDETTE -

Neuromodulation in Experimental Animal Models of Epilepsy
Neuromodulation in Experimental Animal Models of Epilepsy Stefanie Dedeurwaerdere Promoter: Prof. Dr. Paul Boon Co-promoter: Prof. Dr. Walter Verraes Ghent University Neuromodulation in Experimental Animal Models of Epilepsy Stefanie Dedeurwaerdere Thesis submitted to fulfill the requirements for the degree of Doctor in Medical Sciences Promoter: Prof. Dr. Paul Boon Co-promoter: Prof. Dr. Walter Verraes Laboratory for Clinical and Experimental Neurophysiology, Department of Neurology Nothing in life is to be feared It is only to be understood Marie Curie Table of contents Chapter 1: General introduction 5 Chapter 2: General methodology 23 Chapter 3: Efficacy of levetiracetam 43 Dedeurwaerdere,S., Boon,P., De Smedt,T., Claeys,P., Raedt,R., Bosman,T., Van Hese,P., Van Maele,G. and Vonck,K. (2005) Chronic levetiracetam treatment early in life decreases epileptiform events in GAERS, but does not prevent the expression of spike and wave discharges during adulthood. Seizure 14(6), 403-411. Chapter 4: Efficacy of vagus nerve stimulation 61 Dedeurwaerdere,S., Vonck,K., Claeys,P., Van Hese,P., D'Have,M., Grisar,T., Naritoku,D. and Boon,P. (2004). Acute vagus nerve stimulation does not suppress spike and wave discharges in "Genetic Absence Epilepsy Rats from Strasbourg". Epilepsy Res 59, 191-198. Dedeurwaerdere,S., Vonck,K., Van Hese,P., Wadman,W., Boon,P. (2005) The acute and chronic effect of vagus nerve stimulation in "Genetic Absence Epilepsy Rats from Strasbourg" (GAERS). Epilepsia 46(Suppl 5), 94-97. Dedeurwaerdere,S., Gilby,K., Vonck,K., Delbeke,J., Boon,P. and McIntyre,D.C. Vagus nerve stimulation does affect memory in Fast rats, but has both pro-convulsive and anti-convulsive effects on amygdala kindled seizures. -

This Plea Is Not a Ploy to Get More Money to an Underfinanced Sector
--------------------- Gentiana Rosetti Maura Menegatti Franca Camurato Straumann Mai-Britt Schultz Annemie Geerts Doru Jijian Drevariuc Pepa Peneva Barbara Minden Sandro Novosel Mircea Martin Doris Funi Pedro Biscaia Jean-Franois Noville Adina Popescu Natalia Boiadjieva Pyne Frederick Laura Cockett Francisca Van Der Glas Jesper Harvest Marina Torres Naveira Giorgio Baracco Basma El Husseiny Lynn Caroline Brker Louise Blackwell Leslika Iacovidou Ludmila Szewczuk Xenophon Kelsey Renata Zeciene Menndez Agata Cis Silke Kirchhof Antonia Milcheva Elsa Proudhon Barruetabea Dagmar Gester Sophie Bugnon Mathias Lindner Andrew Mac Namara SIGNED BY Zoran Petrovski Cludio Silva Carfagno Jordi Roch Livia Amabilino Claudia Meschiari Elena Silvestri Gioele Pagliaccia Colimard Louise Mihai Iancu Tamara Orozco Ritchie Robertson Caroline Strubbe Stphane Olivier Eliane Bots Florent Perrin Frederick Lamothe Alexandre Andrea Wiarda Robert Julian Kindred Jaume Nadal Nina Jukic Gisela Weimann Mihon Niculescu Laura Alexandra Timofte Nicos Iacovides Maialen Gredilla Boujraf Farida Denise Hennessy-Mills Adolfo Domingo Ouedraogo Antoine D Ivan Gluevi Dilyana Daneva Milena Stagni Fran Mazon Ermis Theodorakis Daniela Demalde’ Adrien Godard Stuart Gill --------------------- Kliment Poposki Maja Kraigher Roger Christmann Andrea-Nartano Anton Merks Katleen Schueremans Daniela Esposito Antoni Donchev Lucy Healy-Kelly Gligor Horia Fernando De Torres Olinka Vitica Vistica Pedro Arroyo Nicolas Ancion Sarunas Surblys Diana Battisti Flesch Eloi Miklos Ambrozy Ian Beavis Mbe -

2010 Credit Union Cherry Blossom Ten Mile Run 1 Men's Results
Men’s Results 2010 Credit Union Cherry Blossom Ten Mile Run Men’s Results 2010 Credit Union Cherry Blossom Ten Mile Run Event Director Message 2010 RACE COMMITTEE Event Director Phil Stewart Deputy Race Director/Treasurer Irv Newman More Good Weather and Plenty of Green Race Support Manager Becky Lambros President Welcome to our first “Green” post-race results publication. As part of our Dennis Steinauer 5K Run-Walk initiative to “Green” this year’s event we have saved some trees by bringing Steve Esmacher the results “book” in electronic format. Enjoy the same pictures, stories and Administration and Scoring Kathy Freedman Men’s Results results as before, but without guilt. Rick Freedman Announcer Let me be the first to offer my congratulations to all of our finishers – you Creigh Kelley Kari Keaton will find your names included in this publication. Awards As always, I am deeply indebted to all of my race committee members Nancy Betress Bag Check – especially Deputy Race Director Irv Newman and Race Support Manager Candice Mothersille Timi Rogers Becky Lambros — who toiled throughout the year to make it all happen. Kevin Everette (5K) In a tough economic environment, our title sponsor, Credit Union Command Central Mark Wheatley Miracle Day, raised over $923,000 for the Children’s Miracle Network. In the Communications eight years of the CUMD’s sponsorship, the event has raised over $4 million Kenny Donovan Consultant dollars to help children in need of medical care through the Children’s Jeff Darman Corrals Miracle Network. A significant portion of those funds goes to Washington, DC’s own Children’s Hospital. -

The Microbiota-Gut-Brain Axis
Physiol Rev 99: 1877–2013, 2019 Published August 28, 2019; doi:10.1152/physrev.00018.2018 THE MICROBIOTA-GUT-BRAIN AXIS John F. Cryan, Kenneth J. O’Riordan, Caitlin S. M. Cowan, Kiran V. Sandhu, Thomaz F. S. Bastiaanssen, Marcus Boehme, Martin G. Codagnone, Sofia Cussotto, Christine Fulling, Anna V. Golubeva, Katherine E. Guzzetta, Minal Jaggar, Caitriona M. Long-Smith, Joshua M. Lyte, Jason A. Martin, Alicia Molinero-Perez, Gerard Moloney, Emanuela Morelli, Enrique Morillas, Rory O’Connor, Joana S. Cruz-Pereira, Veronica L. Peterson, Kieran Rea, Nathaniel L. Ritz, Eoin Sherwin, Simon Spichak, Emily M. Teichman, Marcel van de Wouw, Ana Paula Ventura-Silva, Shauna E. Wallace-Fitzsimons, Niall Hyland, Gerard Clarke, and Timothy G. Dinan APC Microbiome Ireland, University College Cork, Cork, Ireland; Department of Anatomy and Neuroscience, University College Cork, Cork, Ireland; Department of Psychiatry and Neurobehavioural Science, University College Cork, Cork, Ireland; and Department of Physiology, University College Cork, Cork, Ireland Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, Codagnone MG, Cussotto S, Fulling C, Golubeva AV, Guzzetta KE, Jaggar M, Long-Smith CM, Lyte JM, Martin JA, Molinero-Perez A, Moloney G, Morelli E, Morillas E, O’Connor R, Cruz-Pereira JS, Peterson VL, Rea K, Ritz NL, Sherwin E, Spichak S, Teichman EM, van de Wouw M, Ventura-Silva AP, Wallace-Fitzsi- Lmons SE, Hyland N, Clarke G, Dinan TG. The Microbiota-Gut-Brain Axis. Physiol Rev 99: 1877–2013, 2019. Published August 28, 2019; doi:10.1152/physrev.00018.2018.—The importance of the gut-brain axis in maintaining homeostasis has long been appreciated. -

Characterising Gene Regulation During Epileptogenesis in Different Models of Epilepsy
Characterising Gene Regulation during Epileptogenesis in Different Models of Epilepsy Bao-Luen Chang A thesis submitted for the degree of Doctor of Philosophy University College London Department of Clinical and Experimental Epilepsy Institute of Neurology Queen Square 2018 Declaration I, Bao-Luen Chang, confirm that the work presented in this thesis is my original research work. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without the prior written consent of the author. 1 Acknowledgements I would like to express my deepest appreciation to my supervisor Professor Stephanie Schorge for providing me the opportunity to study in the Institute of Neurology, Queen Square. Sincere thanks for her guidance, encouragement and fully support on my research. I am fortunate to meet such a scholar as my role model of a genuine scientist in the research field. I am especially grateful to Professor Matthew Walker and Professor Dimitri Michael Kullmann for their academical expertise, scientific guidance and suggestions on my study. I specially thank Dr. Marife Cano-Jaimez, Dr. Elodie Chabrol, Dr. Gabriele Lignani, and Dr. Pishan Chang for teaching me scientific methods and providing necessary expertise and advice. I am very appreciative to the senior clinical scientist, Dr. James Polke, for his kindly assistance in showing me how to use the gradient PCR and qRT-PCR machines and sharing his experience of experiments using qRT-PCR. I am thankful to all my colleagues in the department of clinical and experimental epilepsy, especially Andrianos Liavas, Albert Snowball, Janosch Heller, Kotaro Sugao, Gareth Morris, Andreas Lieb and Jenna Carpenter, for all their help and friendship. -

UPS Annuario Per L'anno Accademico 2010-2011 LXXI Dalla Fondazione
2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it Università Pontificia Salesiana ANNUARIO PER L’ANNO ACCADEMICO 2010-2011 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it UNIVERSITÀ PONTIFICIA SALESIANA ANNUARIO PER L’ANNO ACCADEMICO 2010-2011 LXXI DALLA FONDAZIONE ROMA 2012 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it Università Pontificia Salesiana Piazza dell’Ateneo Salesiano, 1 00139 Roma - Italia Tel. 06 872901 - Fax 06 87290318 - E-mail: [email protected] Elaborazione elettronica: LAS Stampa: Tip. Abilgraph - Via P. Ottoboni 11 - Roma 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it Presentazione 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it 2015 - Digital Collections - Biblioteca Don Bosco - Roma - http://digital.biblioteca.unisal.it PRESENTAZIONE È ormai diventata una tradizione quella di poter offrire in occasione della fe- sta di don Bosco l’Annuario dell’Università Pontificia Salesiana alle autorità, ai docenti e agli amici dell’Università. Anche quest’anno il volume esce per la generosa disponibilità e laboriosa competenza del prof. Renato Mion, che si è assunto questo non semplice compi- to, malgrado i suoi già abbondanti oneri di docenza e di servizio ecclesiale quale Assistente ecclesiastico dell’Agesc. Dal punto di vista tecnico è affidato al sig. Matteo Cavagnero, operatore dell’editrice universitaria LAS. 1. L’annuario rappresenta il quadro della vita istituzionale dell’Università, espressa attraverso la presentazione della storia, delle strutture, delle persone, delle attività e in qualche modo degli effetti, vale a dire della produzione scien- tifica, culturale e formativa dell’università. -

Saturday, May 12, 2018 Program (PDF)
Spring 2018 • Saturday, May 12 Commencement UNIVERSITY OF WISCONSIN–MADISON UN I V ERSITY OF W ISCONSIN– M A DISON ONE HUNDRED AND SIXTY-FIFTH Commencement Law, Master’s, and Bachelor’s Degrees Saturday, May 12, 2018 12 p.m. Camp Randall Stadium Bascom Hall UNIVERSITY OF WISCONSIN–MADISON One Hundred and Sixty-Fifth Commencement Law, Master’s, and Bachelor’s Degrees Saturday, May 12, 2018 Opening Remarks Master of Music Theresa Andrina La Susa, Master of Professional French Studies Senior Class Vice President, Master of Public Affairs Bachelor of Arts ’18 Master of Science Master of Social Work Wisconsin Medley Dean William J. Karpus, PhD Performed by Redefined, A Capella Remarks on Behalf of the Graduates Processional Ariela Sara Rivkin, Senior Class President, University School of Music Band Bachelor of Arts ’18 Professor Michael Leckrone, MM Wisconsin Alumni Association Video Presentation: Still With You The audience is requested to rise as the procession of officials enters. Recognition of Graduates with Honors and Distinction National Anthem Performed by Katie Anderson Conferral of Baccalaureate Degrees MM, Vocal Performance ’18 College of Agricultural and Life Sciences Welcome on Behalf of the Chancellor Bachelor of Science Chancellor Rebecca M. Blank, PhD Bachelor of Science–Agricultural Business Management Bachelor of Science–Biological Systems Engineering Introduction of the Official Party Bachelor of Science–Dietetics Presiding Officer, Interim Vice Chancellor Bachelor of Science–Landscape Architecture for Student Affairs, Lori M. Berquam, PhD Dean Kathryn A. VandenBosch, PhD Welcome from UW System Board of Regents School of Business Regent Regina Millner, JD Bachelor of Business Administration Charge to the Graduates Interim Dean Barry Gerhart, PhD David Muir School of Education Recognition of Honorary Degree Recipients Bachelor of Fine Arts Honorary Degrees were awarded on Friday, May 11. -

Attendee List
CNU 24 Attendee List First Name Last Name Title Company City State Kate Abbey-Lambertz National Reporter The Huffington Post Detroit Michigan Mark Abell Journalist Buffalo Bibe Buffalo New York Corey Aber Business Design Manager, Community Mission Freddie Mac Multifamily McLean Virginia Daniel Acevedo Urban Design Manager City of Rowlett Rowlett Texas Ken Acton Professor, St Clair College St. Clair College Windsor Ontario shadi adab urban designer M.Arch-OPPI-CIP toronto Ontario jeff Adams Professor of Economics Beloit College Beloit Wisconsin George Adams Executive Director 360 Detroit Inc Detroit Michigan John Adams Community Planning + Revitalization Agency of Commerce and Community Development Montpelier Vermont Alicia Adams Landscape Architect I SmithGroupJJR Ann Arbor Michigan judy Adams Childrens Librarian Beloit Public Library Beloit Wisconsin Kevin Adams Urban Design and Planning Consultant Benchmark Planning Charlotte North Carolina Rik Adamski Principal ASH+LIME Strategies Dallas Texas Charles Adderley Realtor/student Seaman Realty and Management Jacksonville Florida Brian Addison Communications Manager Long Beach California Louis Aguilar The Detroit News Detroit Michigan Hamed Ahangari Student University of Connecticut Vernon Connecticut Khaldoon Ahmad Manager of Urban Design Niagara Region Thorold Ontario John Ahmann Atlanta Committee for Progress Atlanta Georgia Roberta Ahmanson Fieldstead and Company Irvine California Howard Ahmanson President Fieldstead and Company IRVINE California Jonathan Ahn Transportation Planning -

Proquest Dissertations
ALTERTATIONS IN EXCITATORY / INHIBITORY CIRCUITRY IN THE ADULT RAT BRAIN FOLLOWING POSTNATAL GLUTAMATERGIC SYSTEM ACTIVATION BY DAPHNE A. GILL A Thesis Submitted to the Graduate Faculty in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Department of Biomedical Sciences Faculty of Veterinary Medicine University of Prince Edward Island ©2010 D.A. Gill. Library and Archives Bibliothèque et 1^1 Canada Archives Canada Published Heritage Direction du Branch Patrimoine de l’édition 395 Wellington Street 395, rue Wellington Ottawa ON K1A 0N4 Ottawa ON K1A 0N4 Canada Canada Your file Votre référence ISBN: 978 - 0- 494- 64479-9 Our file Notre référence ISBN: 978 - 0- 494- 64479-9 NOTICE: AVIS: The author has granted a non L'auteur a accordé une licence non exclusive exclusive license allowing Library and permettant à la Bibliothèque et Archives Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par télécommunication ou par l’Internet, prêter, telecommunication or on the Internet, distribuer et vendre des thèses partout dans le loan, distribute and sell theses monde, à des fins commerciales ou autres, sur worldwide, for commercial or non support microforme, papier, électronique et/ou commercial purposes, in microform, autres formats. paper, electronic and/or any other formats. The author retains copyright L’auteur conserve la propriété du droit d’auteur ownership and moral rights in this et des droits moraux qui protège cette thèse. Ni thesis. Neither the thesis nor la thèse ni des extraits substantiels de celle-ci substantial extracts from it may be ne doivent être imprimés ou autrement printed or othen/vise reproduced reproduits sans son autorisation. -

11Th European Congress on Epileptology
11TH EUROPEAN CONGRESS ON EPILEPTOLOGY CO N G R E SS P R O G R A M M E STOCKHOLM 201429 JUNE - 3 JULY 1 CONTENTS GENERAL INFORMATION Committees and Welcome Messages 3 Congress Information 6 Congress Centre Layout 10 Stockholm Transport Information 12 Nobel Exhibition 15 SCIENTIFIC PROGRAMME Introductory Message 18 Scientific Programme Information 20 Full Week Timetable 24 Sunday 29th June 26 Monday 30th June 34 Tuesday 1st July 50 Wednesday 2nd July 66 Thursday 3rd July 84 Satellite Symposia 103 POSTERS Poster Information 110 Monday 30th June 119 Tuesday 1st July 138 Wednesday 2nd July 156 OTHER INFORMATION Travel Bursaries 176 Speakers List 177 List of Exhibitors and Floor Plan 190 www.epilepsystockholm2014.org 2 Stockholm 29th June - 3rd July GENERAL INFORMATION GENERAL GENERAL INFORMATION 3 INFORMATION GENERAL GENERAL INFORMATION GENERAL INTERNATIONAL ORGANISING COMMITTEE Co-Chairs: Meir Bialer (Israel) Kristina Malmgren (Sweden) Members: Alla Guekht (Russia) Reetta Kälviäinen (Finland) Ivan Rektor (Czech Republic) Philip Smith (United Kingdom) Torbjörn Tomson (Sweden) SCIENTIFIC ADVISORY COMMITTEE Chair: Torbjörn Tomson (Sweden) Members: Meir Bialer (Israel) Milan Brázdil (Czech Republic) Helen Cross (United Kingdom) Reetta Kälviäinen (Finland) Merab Kokaia (Sweden) Kristina Malmgren (Sweden) Philippe Ryvlin (France) Simon Shorvon (United Kingdom) Federico Vigevano (Italy) www.epilepsystockholm2014.org 4 Stockholm 29th June - 3rd July WELCOME Dear Colleagues, th GENERAL INFORMATION GENERAL On behalf of the International Organising Committee, we wish to welcome you to the ILAE’s 11 European Congress on Epileptology (ECE), in Stockholm. The Stockholm 2014 congress promises to be a very interesting and inspiring congress. The programme will cover cutting-edge research in the field of epilepsy through presentations from the key opinion leaders in the field, including lectures by world-renowned scientists on topics at the interfaces of epilepsy. -
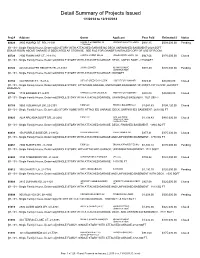
Detail Summary of Projects Issued 1/1/2014 to 12/31/2014
Detail Summary of Projects Issued 1/1/2014 to 12/31/2014 Proj # Address Owner Applicant Fees Paid Estimated $ Status 60624 3805 HUMBOLDT RD..21-391 DANIEL E & KIMBERLY M LEMENSE QUALITY HOMES $992.15 $309,000.00 Pending LIERMANN [01-101 - Single Family House, Detached] 2-STORY WITH ATTACHED GARAGE NO DECK UNFINISHED BASEMENT-2829 SQFT BONUS ROOM ABOVE GARAGE IS DEDICATED AS STORAGE. SEE FILE FOR OWNER'S INITIALED COPY OF USE OF ROOM. 60729 2100 EMMALANE CT..21-8116 CAPITAL CREDIT UNION LEDGECREST HOMES, INC. $947.06 $170,000.00 Closed [01-101 - Single Family House, Detached] SINGLE STORY WITH ATTACHED GARAGE, DECK, UNFIN. BASE. -1786SQFT 60745 425 MCAULIFFE HEIGHTS TR..21-8183 JOHN J BUNKER JOHNNY B HOME $978.39 $170,000.00 Pending CONSTRUCTION [01-101 - Single Family House, Detached] SINGLE STORY WITH ATTACHED GARAGE-1992SQFT 60755 824 GROVE ST..19-47-A CITY OF GREEN BAY-CLERK HABITAT FOR HUMANITY $749.31 $80,000.00 Closed [01-101 - Single Family House, Detached] SINGLE STORY, ATTACHED GARAGE, UNFINISHED BASEMENT-1018SQFT-1ST FLOOR ,264SQFT BASEMENT 60756 1749 BADGER ST..6-570 PATRICK & LYNN LAUGHLIN HABITAT FOR HUMANITY $891.00 $80,000.00 Closed [01-101 - Single Family House, Detached] SINGLE STORY WITH ATTACHED GARAGE, UNFINISHED BASEMENT. 1521 SQ FT 60759 3665 TIDEWATER DR..22-2101 TJDM LLC REINECK BUILDERS LLC $1,041.83 $194,120.00 Closed [01-101 - Single Family House, Detached] 2 STORY HOME WITH ATTACHED GARAGE, DECK, UNFINISHED BASEMENT; 2639 SQ FT 60825 3624 ARCADIA BLUFF DR..22-2082 TJDM LLC NICK HOLTGER $1,708.43 $480,000.00 Closed CONSTRUCTION CORPORATION [01-101 - Single Family House, Detached] SINGLE STORY WITH ATTACHED GARAGE, DECK, FINISHED BASEMENT.