Lawrence Berkeley National Laboratory Recent Work
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Thermal Transport in Thorium Dioxide Nuclear Engineering and Technology
Nuclear Engineering and Technology 50 (2018) 731e737 Contents lists available at ScienceDirect Nuclear Engineering and Technology journal homepage: www.elsevier.com/locate/net Original Article Thermal transport in thorium dioxide Jungkyu Park*, Eduardo B. Farfan, Christian Enriquez Kennesaw State University, Department of Mechanical Engineering, Kennesaw, GA 30144, USA article info abstract Article history: In this research paper, the thermal transport in thorium dioxide is investigated by using nonequilibrium Received 28 September 2017 molecular dynamics. The thermal conductivity of bulk thorium dioxide was measured to be 20.8 W/m-K, Received in revised form confirming reported values, and the phonon mean free path was estimated to be between 7 and 8.5 nm 20 December 2017 at 300 K. It was observed that the thermal conductivity of thorium dioxide shows a strong dependency Accepted 12 February 2018 on temperature; the highest thermal conductivity was estimated to be 77.3 W/m-K at 100 K, and the Available online 1 March 2018 lowest thermal conductivity was estimated to be 4.3 W/m-K at 1200 K. In addition, by simulating thorium dioxide structures with different lengths at different temperatures, it was identified that short Keywords: Molecular Dynamics Simulation wavelength phonons dominate thermal transport in thorium dioxide at high temperatures, resulting in Nuclear Fuel decreased intrinsic phonon mean free paths and minimal effect of boundary scattering while long Thermal Transport wavelength phonons dominate the thermal transport in thorium dioxide at low temperatures. Thorium Dioxide © 2018 Korean Nuclear Society, Published by Elsevier Korea LLC. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). -

INFORMATION O a a .Aoi Chemistry-Traniuranie Elements
INFORMATION o a a .aoi Chemistry-Traniuranie Elements UNIVERSITY O r CALIFORNIA Radiation Laboratory Contract No. W-7405-eng~48 CUBSITIClTIOH CAKCBLUD IV. TT Ftv«Jbs Itti'.s fJ«cR'otsdLisV^ CRYSTAL STRUCTURES OF AMERICIUM COMPOUNDS D. H. Templeton and Carol H. Dauben February 3, 1953 RESTRICTEO^JCTA This document cqpm pLrw ricted data as defined in ttagSrarfic Energy Act of 1944. Its tsn fip rtfa l or disclosure of Its contents ^p*ny manner to an unautor- ised person is prohibited. Berkeley, California UNIVERSITY Of CALIFORNIA Rodiotiofl Loborotory 42RC8 Cover Shoot INDE X NO. Ill K£ ■ -10/ Do not rsrtovt Tbit document eo nto in t__ IL- pages Tbit is copy ^ of tor lot tL Glassification Eoch perton who rtcoivot thltdocument must tipn the cover thstt I Route to Notsd by Date Route to Noted by D aft OHO A im *[|S 5(M) CRYSTAL STRUCTURES OF AMERICIUM COMPOUNDS1 D. H. Templeton and Carol H. Dauben Department of Chemistry and Radiation Laboratory University of California, Berkeley, California February 3, 1953 ABSTRACT The crystal structures of several compounds of americium, element 95, have been determined by the X-ray powder diffraction method. AisF^ is hexagonal (LaF^ type) with a = 4. 067 t 0.001 X and c « 7. 225 t 0.002 Si for the pseudoceil which explains the powder data. AmO^ is cubic (CaF^ type) with 1 a = 5. 38) t 0.001 X AmOCl is tetragonal (PbFCl type) with a = 4.00+O.OlX, c = 6.78 t 0.01 X. The metal parameter la 0.18 t 0.01. -

Subject Index
SUBJECT INDEX Vol. 1: 1–698, Vol. 2: 699–1395, Vol. 3: 1397–2111, Vol. 4: 2113–2798, Vol. 5: 2799–3440. Page numbers suffixed by t and f refer to Tables and Figures respectively. Absorption spectra in aqueous solution, 752–770 of americium, 1364–1368 compounds of, 721–752 americium (III), 1364–1365, 1365f coordination complexes in solution, americium (IV), 1365 771–782 americium (V), 1366, 1367f history of, 699–700 americium (VI), 1366, 1367f isotope production, 702–703 americium (VII), 1367–1368, 1368f metallic state of, 717–721 of liquid plutonium, 963 in nature, 703–704 of neptunium, 763–766, 763f, 786–787 nuclear properties of, 700–702 neptunium (VII) ternary oxides, 729 separation and purification, 704–717 of plutonium plutonium hexafluoride, 1088, 1089f atomic properties of, 857–862 ions, 1113–1117, 1116t compounds of, 987–1108 plutonium (IV), 849 metal and intermetallic compounds of, polymerization, 1151, 1151f 862–987 tetrachloride, 1093–1094, 1094f natural occurrence of, 822–824 tribromide, 1099t, 1100 nuclear properties of, 815–822 trichloride, 1099, 1099t separation and purification of, 826–857 Accelerator mass spectrometry (AMS), of solution chemistry of, 1108–1203 neptunium, 790 Actinide elements Acetates atomic volumes of, 922–923, 923f of americium, 1322, 1323t extraction of of plutonium, 1177, 1180 DIDPA, 1276 Acetone, derived compounds, of americium, HDEHP, 1275 1322, 1323t, 1324 organophosphorus and Acids carbamoylphosphonate reagents, for Purex process, 711 1276–1278 for solvent extraction, 839 stripping of, 1280–1281 Actinide -

Electrical Characterization of Crystalline UO 2 , Tho 2 and U
Air Force Institute of Technology AFIT Scholar Theses and Dissertations Student Graduate Works 12-17-2018 Electrical Characterization of Crystalline UO2, ThO2 AND U0.71Th0.29O2 Christina L. Dugan Follow this and additional works at: https://scholar.afit.edu/etd Part of the Engineering Physics Commons Recommended Citation Dugan, Christina L., "Electrical Characterization of Crystalline UO2, ThO2 AND U0.71Th0.29O2" (2018). Theses and Dissertations. 1944. https://scholar.afit.edu/etd/1944 This Dissertation is brought to you for free and open access by the Student Graduate Works at AFIT Scholar. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of AFIT Scholar. For more information, please contact [email protected]. ELECTRICAL CHARACTERIZATION OF CRYSTALLINE UO2, THO2 AND U0.71TH0.29O2 DISSERTATION Christina L. Dugan, Lieutenant Colonel, USA AFIT-ENP-DS-18-D-007 DEPARTMENT OF THE AIR FORCE AIR UNIVERSITY AIR FORCE INSTITUTE OF TECHNOLOGY Wright-Patterson Air Force Base, Ohio DISTRIBUTION STATEMENT A Approved For Public Release; Distribution Unlimited The views expressed in this thesis are those of the author and do not reflect the official policy or position of the United States Air Force, the Department of Defense, or the United States Government. AFIT-ENP-DS-18-D-007 ELECTRICAL CHARACTERIZATION OF CRYSTALLINE UO2, THO2 AND U0.71TH0.29O2 DISSERTATION Presented to the Faculty Department of Engineering Physics Graduate School of Engineering and Management Air Force Institute of Technology Air University Air Education and Training Command In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Christina L. -

Plutonium and Americium Geochemistry at Hanford
Plutonium Futures: The Science 2014 Las Vegas, Nevada, USA 7-12 September 2014 ISBN: 978-1-5108-0808-9 Printed from e-media with permission by: Curran Associates, Inc. 57 Morehouse Lane Red Hook, NY 12571 Some format issues inherent in the e-media version may also appear in this print version. Copyright© (2014) by the American Nuclear Society All rights reserved. Printed by Curran Associates, Inc. (2015) For permission requests, please contact the American Nuclear Society at the address below. American Nuclear Society 555 North Kensington Avenue LaGrange Park, Illinois 60526 Phone: (800) 323-3044 (708) 352-6611 Fax: (708) 352-0499 www.ans.org Additional copies of this publication are available from: Curran Associates, Inc. 57 Morehouse Lane Red Hook, NY 12571 USA Phone: 845-758-0400 Fax: 845-758-2634 Email: [email protected] Web: www.proceedings.com Table of Contents MONDAY, SEPTEMBER 8, 2014, 8:00-10:20 A.M. MONDAY PLENARIES Session Chair: Kerri Blobaum (LLNL) 1Plutonium and Americium Geochemistry at Hanford A. R. Felmy, K. J. Cantrell, E. C. Buck (PNNL), S. D. Conradson (LANL), plenary 3Consequences of Electronic Correlations in Plutonium-Based Intermetallics J. D. Thompson, E. D. Bauer, G. Koutroulakis, J. N. Mitchell, A. M. Mounce, P. H. Tobash, H. Yasuoka (LANL), plenary MONDAY, SEPTEMBER 8, 2014, 10:40 A.M-12:00 P.M. CONDENSED MATTER PHYSICS—I Session Chair: Roberto Caciuffo (ITU) 5A Plutonium-Based Single-Molecule Magnet N. Magnani, E. Colineau, J.-C. Griveau, C. Apostolidis, O. Walter, R. Caciuffo (EC, JRC, Inst for Transuranium Elements), invited 6Quantum Criticality in PuMX5(M=Co, Rh; X=Ga, In) Superconductors Eric D. -

Periodic Table of Elements
The origin of the elements – Dr. Ille C. Gebeshuber, www.ille.com – Vienna, March 2007 The origin of the elements Univ.-Ass. Dipl.-Ing. Dr. techn. Ille C. Gebeshuber Institut für Allgemeine Physik Technische Universität Wien Wiedner Hauptstrasse 8-10/134 1040 Wien Tel. +43 1 58801 13436 FAX: +43 1 58801 13499 Internet: http://www.ille.com/ © 2007 © Photographs of the elements: Mag. Jürgen Bauer, http://www.smart-elements.com 1 The origin of the elements – Dr. Ille C. Gebeshuber, www.ille.com – Vienna, March 2007 I. The Periodic table............................................................................................................... 5 Arrangement........................................................................................................................... 5 Periodicity of chemical properties.......................................................................................... 6 Groups and periods............................................................................................................. 6 Periodic trends of groups.................................................................................................... 6 Periodic trends of periods................................................................................................... 7 Examples ................................................................................................................................ 7 Noble gases ....................................................................................................................... -

Thesis Petronela Gotcu 15 ... FINAL.Pdf
High temperature thermodynamic studies on the transuranium oxides and their solid solutions Proefschrift ter verkrijging van de graad van doctor aan de Technische Universiteit Delft, op gezag van de Rector Magnificus Prof.ir. K.C.A.M. Luyben, voorzitter van het College voor Promoties, in het openbaar te verdedigen op woensdag 15 juni 2011 om 10.00 uur door Petronela GOTCU-FREIS Magister in de chemie van de Faculteit Chemie van de „Alexandru Ioan Cuza” Universiteit van Iaşi, Roemenië geboren te Iaşi, Roemenië Dit proefschrift is goedgekeurd door de promotoren: Prof.dr. H.Th. Wolterbeek Prof.dr. R.J.M. Konings Samenstelling promotiecommissie: Rector Magnificus, voorzitter Prof.dr. H.Th. Wolterbeek, Technische Universiteit Delft, promotor Prof.dr. R.J.M. Konings, Technische Universiteit Delft, promotor Prof.dr.ir. A. Verkooijen, Technische Universiteit Delft Prof.dr.ir. E. Bruck, Technische Universiteit Delft Prof.dr. K. Hack, RWTH Aachen University, Duitsland Prof.dr. D. Haas, VU Brussels, België Dr. Ch. Guéneau, CEA Saclay, Frankrijk Prof.dr.ir. C. Pappas, Technische Universiteit Delft, reservelid The research described in this thesis was performed within a cooperation of the Institute of Transuranium Elements of the Joint Research Centre (JRC) of the European Commission, P.O. Box 2340, 76125 Karlsruhe, Germany and the Section of Radiation and Isotopes for Health of the Department of Radiation, Radionuclides and Reactors, Faculty of Applied Sciences, Delft University of Technology, Melkeweg 15, 2629 JB Delft, The Netherlands. © 2011 P. Gotcu-Freis and IOS Press All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without prior permission from the publisher. -

HYSYS OLI Interface
HYSYS® 2004.2 OLI Interface Reference Guide Copyright October 2005 Copyright © 1981-2005 by Aspen Technology, Inc. All rights reserved. Aspen Accounting.21™, Aspen ACM Model Export, Aspen ACOL™, Aspen ACX™ Upgrade to ACOL™, Aspen Adsim®, Aspen Advisor™, Aspen Aerotran®, Aspen Alarm & Event™, Aspen APLE™, Aspen Apollo™, Aspen AtOMS™, Aspen Batch and Event Extractor, Aspen Batch Plus®, Aspen Batch.21™, Aspen Batch.21™ CBT, Aspen BatchCAD™, Aspen BatchSep™, Aspen Blend Model Library™, Aspen Blend™, Aspen BP Crude Oil Database, Aspen Calc CBT, Aspen Calc™, Aspen Capable-to-Promise®, Aspen CatRef®, Aspen Chromatography®, Aspen Cim-IO Core™, Aspen Cim-IO™ for @AGlance, Aspen Cim-IO™ for ABB 1180/ 1190 via DIU, Aspen Cim-IO™ for Bailey SemAPI, Aspen Cim-IO™ for DDE, Aspen Cim-IO™ for Eurotherm Gauge via DCP, Aspen Cim-IO™ for Fisher-Rosemount Chip, Aspen Cim-IO™ for Fisher-Rosemount RNI, Aspen Cim-IO™ for Foxboro FOXAPI, Aspen Cim-IO™ for G2, Aspen Cim-IO™ for GE FANUC via HCT, Aspen Cim-IO™ for Hitachi Ex Series, Aspen Cim-IO™ for Honeywell TDC 3000 via HTL/access, Aspen Cim-IO™ for Intellution Fix, Aspen Cim-IO™ for Measurex MCN, Aspen Cim-IO™ for Measurex ODX, Aspen Cim-IO™ for Moore Apacs via Nim (RNI), Aspen Cim-IO™ for OPC, Aspen Cim-IO™ for PI, Aspen Cim- IO™ for RSLinx, Aspen Cim-IO™ for SetCim/InfoPlus-X/InfoPlus.21, Aspen Cim-IO™ for Toshiba Tosdic, Aspen Cim-IO™ for ULMA 3D, Aspen Cim-IO™ for Westinghouse, Aspen Cim-IO™ for WonderWare InTouch, Aspen Cim-IO™ for Yokogawa ACG10S, Aspen Cim-IO™ for Yokogawa EW3, Aspen Collaborative Forecasting™, -

The European Institute for Transuranium Element
Commission of the European Communities ·-·-· Research Centre The European Institute for Transuranium Element The European Institute for Transuranium Elements Published by the COMMISSION OF THE EUROPEAN COMMUNITIES Directorate-CJeneral Information Market and Innovation Bat1ment Jean Monnet LUXEMBOURG LEGAL NOTICE Neither the Comm1ss1on of 1he European Commun1t1es nor any person act1ng on behalf of the CommiSSion IS responsible for the use wh1ch m1ght be made of the following information Cataloguing data can be found at the end of th1s publ1cat1on Luxembourg, Off1ce for OffiCial Publications of the European Commumt1es 1986 ISBN 92-825-6100-3 Catalogue number CD-NE-86-018-EN-C © ECSC-EEC-EAEC, Brussels · Luxembourg, 1986 Prmted m the Federal Republtc of Germany , THE EUROPEAN INSTITUTE FOR TRANSURANIUM ELEMENTS Commission of the European Communities Joint Research Centre (JRC) Karlsruhe Establishment Directorate-General Science, Research and Development 1986 EUR 10480 EN Contents Adress from the Outgoing Director Adress from the Incoming Director 1 At a Glance 11 Introduction 11 What are Transuranium Elements? 12 The Institute 14 The Programme 17 2 The Mission 21 2.1 Background Information 21 The European Communities and their Treaties 21 The EURATOM Treaty The Transuranium Elements 22 - Formation of Transuranium Elements - Nuclear Stability and Superheavy Elements - The Actinide Concept of Transuranium Elements - Application of Transuranium Elements The Transuranium Institute 27 - Some Historic Dates and Facts - Personnel -Buildings - Technical -
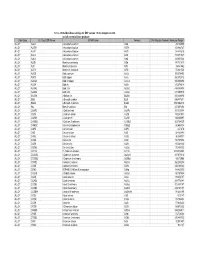
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

The Transuranium Elements
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title The Transuranium Elements Permalink https://escholarship.org/uc/item/56h2r51f Authors Hyde, Earl K. Seaborg, Glenn T. Publication Date 1956-02-17 eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Radiation Laboratory Berkeley, California Contract No. W-7405-eng-48 THE TRANSURANIUM ELEMENTS Earl K. Hyde and Glenn T. Seaborg February 17, 1956 .Preprint of a contribution to Volume 39, "Handbuch der Physik" Printed for the U. S. Atomic Energy Commission THE TRANSURANPUM ELEMENTS Earl K. Hyde and Glenn T. Seabor g OUT LINE A. GENERAL CONSIDERATIONS - THE PLACE OF THE HEAVIEST ELEMENTS IN THE PERIODIC SYSTEM I. HISTORICAL VIEWS 1. Speculations previous to discovery of transuranium elements 2. The actinide concept. 11. EVIDENCE RELATING TO THE ACTINIDE CONCEPT a. Chemical Evidence 3. Tripositive oxidation state in aqueous solution 4. Stability of anhydrous halides 5. Discussion of evidence on oxidation states 6. Crystallographic data--isostructural series 7. The metallic state 8. Ion-exchange behavior b. Pbys ical Evidence 9. Absorption and fluorescence spectra in aqueous solution and crystals 10. Magnetic susceptibility data - theoretical 11, Magnetic susceptibility data - experimental studies 12. Spectroscopic data B. THE INDIVIDUAL ELEMENTS I. NEPTUNIUM (ELEMENT 93) a. History of Neptunium 13. The transuranium elements before the discovery of fission 14. Discovery of neptunium - summary of isotopes b. Chemical Properties of Neptunium 15. The oxidation states 16. Absorption spectra 17. Neptunium(II1) in aqueous solution 18. Neptunium(1V) in aqueous s olution 19. NeptuniumBV) in aqueous solution 20. Neptunium(V1) in aqueous solution 21. -

Speciation Behavior of Americium Higher Oxidation States for the Separation of Americium from Curium
UNLV Theses, Dissertations, Professional Papers, and Capstones 8-1-2014 Speciation Behavior of Americium Higher Oxidation States for the Separation of Americium from Curium Catherine Lynn Riddle University of Nevada, Las Vegas Follow this and additional works at: https://digitalscholarship.unlv.edu/thesesdissertations Part of the Oil, Gas, and Energy Commons, and the Radiochemistry Commons Repository Citation Riddle, Catherine Lynn, "Speciation Behavior of Americium Higher Oxidation States for the Separation of Americium from Curium" (2014). UNLV Theses, Dissertations, Professional Papers, and Capstones. 2208. http://dx.doi.org/10.34917/6456439 This Dissertation is protected by copyright and/or related rights. It has been brought to you by Digital Scholarship@UNLV with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/or on the work itself. This Dissertation has been accepted for inclusion in UNLV Theses, Dissertations, Professional Papers, and Capstones by an authorized administrator of Digital Scholarship@UNLV. For more information, please contact [email protected]. SPECIATION BEHAVIOR OF AMERICIUM HIGHER OXIDATION STATES FOR THE SEPARATION OF AMERICIUM FROM CURIUM by Catherine Lynn Riddle Bachelor of Science in Chemistry Idaho State