Speciation Behavior of Americium Higher Oxidation States for the Separation of Americium from Curium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermal Transport in Thorium Dioxide Nuclear Engineering and Technology
Nuclear Engineering and Technology 50 (2018) 731e737 Contents lists available at ScienceDirect Nuclear Engineering and Technology journal homepage: www.elsevier.com/locate/net Original Article Thermal transport in thorium dioxide Jungkyu Park*, Eduardo B. Farfan, Christian Enriquez Kennesaw State University, Department of Mechanical Engineering, Kennesaw, GA 30144, USA article info abstract Article history: In this research paper, the thermal transport in thorium dioxide is investigated by using nonequilibrium Received 28 September 2017 molecular dynamics. The thermal conductivity of bulk thorium dioxide was measured to be 20.8 W/m-K, Received in revised form confirming reported values, and the phonon mean free path was estimated to be between 7 and 8.5 nm 20 December 2017 at 300 K. It was observed that the thermal conductivity of thorium dioxide shows a strong dependency Accepted 12 February 2018 on temperature; the highest thermal conductivity was estimated to be 77.3 W/m-K at 100 K, and the Available online 1 March 2018 lowest thermal conductivity was estimated to be 4.3 W/m-K at 1200 K. In addition, by simulating thorium dioxide structures with different lengths at different temperatures, it was identified that short Keywords: Molecular Dynamics Simulation wavelength phonons dominate thermal transport in thorium dioxide at high temperatures, resulting in Nuclear Fuel decreased intrinsic phonon mean free paths and minimal effect of boundary scattering while long Thermal Transport wavelength phonons dominate the thermal transport in thorium dioxide at low temperatures. Thorium Dioxide © 2018 Korean Nuclear Society, Published by Elsevier Korea LLC. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). -

A Protein Engineered to Bind Uranyl Selectively and with Femtomolar Affinity
ARTICLES PUBLISHED ONLINE: 26 JANUARY 2014 | DOI: 10.1038/NCHEM.1856 A protein engineered to bind uranyl selectively and with femtomolar affinity Lu Zhou1†, Mike Bosscher1†, Changsheng Zhang2,3†,SalihO¨ zc¸ubukc¸u1, Liang Zhang1, Wen Zhang1, Charles J. Li1, Jianzhao Liu1, Mark P. Jensen4, Luhua Lai2,3* and Chuan He1* 21 ∼ Uranyl (UO2 ), the predominant aerobic form of uranium, is present in the ocean at a concentration of 3.2 parts per 109 (13.7 nM); however, the successful enrichment of uranyl from this vast resource has been limited by the high concentrations of metal ions of similar size and charge, which makes it difficult to design a binding motif that is selective for uranyl. Here we report the design and rational development of a uranyl-binding protein using a computational screening process in the initial search for potential uranyl-binding sites. The engineered protein is thermally stable and offers very high affinity and selectivity for uranyl with a Kd of 7.4 femtomolar (fM) and >10,000-fold selectivity over other metal ions. We also demonstrated that the uranyl-binding protein can repeatedly sequester 30–60% of the uranyl in synthetic sea water. The chemical strategy employed here may be applied to engineer other selective metal-binding proteins for biotechnology and remediation applications. ranium is the key element for nuclear-energy production and Through billions of years of evolution nature has produced is important in many other applications. The most stable and strategies to recognize beneficial or toxic metal ions with high Urelevant uranium ion in aerobic environments is the uranyl sensitivity and selectivity. -

Hierarchically-Structured Metalloprotein Composite
ARTICLE There are amendments to this paper https://doi.org/10.1038/s41467-020-14709-y OPEN Hierarchically-structured metalloprotein composite coatings biofabricated from co-existing condensed liquid phases ✉ Franziska Jehle 1, Elena Macías-Sánchez1, Sanja Sviben1, Peter Fratzl1, Luca Bertinetti 1 & ✉ Matthew J. Harrington 1,2 1234567890():,; Complex hierarchical structure governs emergent properties in biopolymeric materials; yet, the material processing involved remains poorly understood. Here, we investigated the multi- scale structure and composition of the mussel byssus cuticle before, during and after for- mation to gain insight into the processing of this hard, yet extensible metal cross-linked protein composite. Our findings reveal that the granular substructure crucial to the cuticle’s function as a wear-resistant coating of an extensible polymer fiber is pre-organized in con- densed liquid phase secretory vesicles. These are phase-separated into DOPA-rich proto- granules enveloped in a sulfur-rich proto-matrix which fuses during secretion, forming the sub-structure of the cuticle. Metal ions are added subsequently in a site-specific way, with iron contained in the sulfur-rich matrix and vanadium coordinated by DOPA-catechol in the granule. We posit that this hierarchical structure self-organizes via phase separation of specific amphiphilic proteins within secretory vesicles, resulting in a meso-scale structuring that governs cuticle function. 1 Department of Biomaterials, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, -

Crystal Structure of a Material Is Way in Which Atoms, Ions, Molecules Are Spatially Arranged in 3-D Space
Crystalline Structures – The Basics •Crystal structure of a material is way in which atoms, ions, molecules are spatially arranged in 3-D space. •Crystal structure = lattice (unit cell geometry) + basis (atom, ion, or molecule positions placed on lattice points within the unit cell). •A lattice is used in context when describing crystalline structures, means a 3-D array of points in space. Every lattice point must have identical surroundings. •Unit cell: smallest repetitive volume •Each crystal structure is built by stacking which contains the complete lattice unit cells and placing objects (motifs, pattern of a crystal. A unit cell is chosen basis) on the lattice points: to represent the highest level of geometric symmetry of the crystal structure. It’s the basic structural unit or building block of crystal structure. 7 crystal systems in 3-D 14 crystal lattices in 3-D a, b, and c are the lattice constants 1 a, b, g are the interaxial angles Metallic Crystal Structures (the simplest) •Recall, that a) coulombic attraction between delocalized valence electrons and positively charged cores is isotropic (non-directional), b) typically, only one element is present, so all atomic radii are the same, c) nearest neighbor distances tend to be small, and d) electron cloud shields cores from each other. •For these reasons, metallic bonding leads to close packed, dense crystal structures that maximize space filling and coordination number (number of nearest neighbors). •Most elemental metals crystallize in the FCC (face-centered cubic), BCC (body-centered cubic, or HCP (hexagonal close packed) structures: Room temperature crystal structure Crystal structure just before it melts 2 Recall: Simple Cubic (SC) Structure • Rare due to low packing density (only a-Po has this structure) • Close-packed directions are cube edges. -

Royal Society of Chemistry Input to the Ad Hoc Nuclear
ROYAL SOCIETY OF CHEMISTRY INPUT TO THE AD HOC NUCLEAR RESEARCH AND DEVELOPMENT ADVISORY BOARD The Royal Society of Chemistry (RSC) was pleased to hear of the instigation of the Ad Hoc Nuclear Research and Development Advisory Board (the Board) following the findings of the House of Lords Science and Technology Committee Inquiry ‘Nuclear Research and Development Capabilities’.1,2 The RSC is the largest organisation in Europe for advancing the chemical sciences. Supported by a network of 47,000 members worldwide and an internationally acclaimed publishing business, its activities span education and training, conferences and science policy, and the promotion of the chemical sciences to the public. This document represents the views of the RSC. The RSC has a duty under its Royal Charter "to serve the public interest" by acting in an independent advisory capacity, and it is in this spirit that this submission is made. To provide input to the Board the RSC has performed a wide consultation with the chemical science community, including members of both our Radiochemistry and Energy Sector Interest Groups and also our Environment Sustainability and Energy Division. September 2012 The Role of Chemistry in a Civil Nuclear Strategy 1 Introduction Chemistry and chemical knowledge is essential in nuclear power generation and nuclear waste management. It is essential that a UK civil nuclear strategy recognises the crucial role that chemistry plays, both in research and innovation and in the development of a strong skills pipeline. As the RSC previously articulated in our response to the House of Lords Inquiry, 3 nuclear power is an important component of our current energy mix. -

Types of Lattices
Types of Lattices Types of Lattices Lattices are either: 1. Primitive (or Simple): one lattice point per unit cell. 2. Non-primitive, (or Multiple) e.g. double, triple, etc.: more than one lattice point per unit cell. Double r2 cell r1 r2 Triple r1 cell r2 r1 Primitive cell N + e 4 Ne = number of lattice points on cell edges (shared by 4 cells) •When repeated by successive translations e =edge reproduce periodic pattern. •Multiple cells are usually selected to make obvious the higher symmetry (usually rotational symmetry) that is possessed by the 1 lattice, which may not be immediately evident from primitive cell. Lattice Points- Review 2 Arrangement of Lattice Points 3 Arrangement of Lattice Points (continued) •These are known as the basis vectors, which we will come back to. •These are not translation vectors (R) since they have non- integer values. The complexity of the system depends upon the symmetry requirements (is it lost or maintained?) by applying the symmetry operations (rotation, reflection, inversion and translation). 4 The Five 2-D Bravais Lattices •From the previous definitions of the four 2-D and seven 3-D crystal systems, we know that there are four and seven primitive unit cells (with 1 lattice point/unit cell), respectively. •We can then ask: can we add additional lattice points to the primitive lattices (or nets), in such a way that we still have a lattice (net) belonging to the same crystal system (with symmetry requirements)? •First illustrate this for 2-D nets, where we know that the surroundings of each lattice point must be identical. -

Depleted Uranium Technical Brief
Disclaimer - For assistance accessing this document or additional information,please contact [email protected]. Depleted Uranium Technical Brief United States Office of Air and Radiation EPA-402-R-06-011 Environmental Protection Agency Washington, DC 20460 December 2006 Depleted Uranium Technical Brief EPA 402-R-06-011 December 2006 Project Officer Brian Littleton U.S. Environmental Protection Agency Office of Radiation and Indoor Air Radiation Protection Division ii iii FOREWARD The Depleted Uranium Technical Brief is designed to convey available information and knowledge about depleted uranium to EPA Remedial Project Managers, On-Scene Coordinators, contractors, and other Agency managers involved with the remediation of sites contaminated with this material. It addresses relative questions regarding the chemical and radiological health concerns involved with depleted uranium in the environment. This technical brief was developed to address the common misconception that depleted uranium represents only a radiological health hazard. It provides accepted data and references to additional sources for both the radiological and chemical characteristics, health risk as well as references for both the monitoring and measurement and applicable treatment techniques for depleted uranium. Please Note: This document has been changed from the original publication dated December 2006. This version corrects references in Appendix 1 that improperly identified the content of Appendix 3 and Appendix 4. The document also clarifies the content of Appendix 4. iv Acknowledgments This technical bulletin is based, in part, on an engineering bulletin that was prepared by the U.S. Environmental Protection Agency, Office of Radiation and Indoor Air (ORIA), with the assistance of Trinity Engineering Associates, Inc. -

The Metrical Matrix in Teaching Mineralogy G. V
THE METRICAL MATRIX IN TEACHING MINERALOGY G. V. GIBBS Department of Geological Sciences & Department of Materials Science and Engineering Virginia Polytechnic Institute and State University Blacksburg, VA 24061 [email protected] INTRODUCTION The calculation of the d-spacings, the angles between planes and zones, the bond lengths and angles and other important geometric relationships for a mineral can be a tedious task both for the student and the instructor, particularly when completed with the large as- sortment of trigonometric identities and algebraic formulae that are available (d. Crystal Geometry (1959), Donnay and Donnay, International Tables for Crystallography, Vol. II, Section 3, The Kynoch Press, 101-158). However, such calculations are straightforward and relatively easy to do when completed with the metrical matrix and the interactive software MATOP. Several applications of the matrix are presented below, each of which is worked out in detail and which is designed to teach you its use in the study of crystal geometry. SOME PRELIMINARY COMMENTS We begin our discussion of the matrix with a brief examination of the properties of the geometric three dimensional space, S, in which we live and in which minerals and rocks occur. For our purposes, it will be convenient to view S as the set of all vectors that radiate from a common origin to each point in space. In constructing a model for S, we chose three noncoplanar, coordinate axes denoted X, Y and Z, each radiating from the origin, O. Next, we place three nonzero vectors denoted a, band c along X, Y and Z, respectively, likewise radiating from O. -
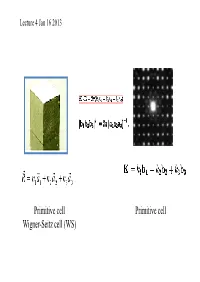
Primitive Cell Wigner-Seitz Cell (WS) Primitive Cell
Lecture 4 Jan 16 2013 Primitive cell Primitive cell Wigner-Seitz cell (WS) First Brillouin zone The Wigner-Seitz primitive cell of the reciprocal lattice is known as the first Brillouin zone. (Wigner-Seitz is real space concept while Brillouin zone is a reciprocal space idea). Powder cell Polymorphic Forms of Carbon Graphite – a soft, black, flaky solid, with a layered structure – parallel hexagonal arrays of carbon atoms – weak van der Waal’s forces between layers – planes slide easily over one another Miller indices Simple cubic Miller Indices Rules for determining Miller Indices: 1. Determine the intercepts of the face along the crystallographic axes, in terms of un it ce ll dimens ions. 2. Take the reciprocals 3. Clear fractions 4. Reduce to lowest terms Simple cubic d100=? Where does a protein crystallographer see the Miller indices? CtlCommon crystal faces are parallel to lattice planes • Eac h diffrac tion spo t can be regarded as a X-ray beam reflected from a lattice plane , and therefore has a unique Miller index. Miller indices A Miller index is a series of coprime integers that are inversely ppproportional to the intercepts of the cry stal face or crystallographic planes with the edges of the unit cell. It describes the orientation of a plane in the 3-D lattice with respect to the axes. The general form of the Miller index is (h, k, l) where h, k, and l are integers related to the unit cell along the a, b, c crystal axes. Irreducible brillouin zone II II II II Reciprocal lattice ghakblc The Bravais lattice after Fourier transform real space reciprocal lattice normaltthlls to the planes (vect ors ) poitints spacing between planes 1/distance between points ((y,p)actually, 2p/distance) l (distance, wavelength) 2p/l=k (momentum, wave number) BillBravais cell Wigner-SiSeitz ce ll Brillouin zone . -
![A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4n]3[UO2(Ncse)5]](https://docslib.b-cdn.net/cover/7628/a-structural-and-spectroscopic-study-of-the-first-uranyl-selenocyanate-et4n-3-uo2-ncse-5-307628.webp)
A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4n]3[UO2(Ncse)5]
inorganics Article A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5] Stefano Nuzzo, Michelle P. Browne, Brendan Twamley, Michael E. G. Lyons and Robert J. Baker * School of Chemistry, Trinity College, University of Dublin, 2 Dublin, Ireland; [email protected] (S.N.); [email protected] (M.P.B.); [email protected] (B.T.); [email protected] (M.E.G.L.) * Correspondence: [email protected]; Tel.: +353-1-896-3501; Fax: +353-1-671-2826 Academic Editors: Stephen Mansell and Steve Liddle Received: 30 October 2015; Accepted: 4 February 2016; Published: 16 February 2016 Abstract: The first example of a uranyl selenocyanate compound is reported. The compound [Et4N]3[UO2(NCSe)5] has been synthesized and fully characterized by vibrational and multinuclear (1H, 13C{1H} and 77Se{1H}) NMR spectroscopy. The photophysical properties have also been recorded and trends in a series of uranyl pseudohalides discussed. Spectroscopic evidence shows that the U–NCSe bonding is principally ionic. An electrochemical study revealed that the reduced uranyl(V) species is unstable to disproportionation and a ligand based oxidation is also observed. The structure of [Et4N]4[UO2(NCSe)5][NCSe] is also presented and Se¨ ¨ ¨ H–C hydrogen bonding and Se¨ ¨ ¨ Se chalcogen–chalcogen interactions are seen. Keywords: uranyl; structural determination; photophysics 1. Introduction The chemistry of uranium in its highest oxidation state has held scientists fascination for a long 2+ period of time. The uranyl moiety, [UO2] , is well studied in aqueous phases due, in part, to relevance in the nuclear waste treatment. Moreover, the photophysical properties of uranyl were first used in ancient roman times in colored glass [1], whilst comprehensive understanding of the bonding, and therefore photophysical properties, has come from both experiment and theory. -

INFORMATION O a a .Aoi Chemistry-Traniuranie Elements
INFORMATION o a a .aoi Chemistry-Traniuranie Elements UNIVERSITY O r CALIFORNIA Radiation Laboratory Contract No. W-7405-eng~48 CUBSITIClTIOH CAKCBLUD IV. TT Ftv«Jbs Itti'.s fJ«cR'otsdLisV^ CRYSTAL STRUCTURES OF AMERICIUM COMPOUNDS D. H. Templeton and Carol H. Dauben February 3, 1953 RESTRICTEO^JCTA This document cqpm pLrw ricted data as defined in ttagSrarfic Energy Act of 1944. Its tsn fip rtfa l or disclosure of Its contents ^p*ny manner to an unautor- ised person is prohibited. Berkeley, California UNIVERSITY Of CALIFORNIA Rodiotiofl Loborotory 42RC8 Cover Shoot INDE X NO. Ill K£ ■ -10/ Do not rsrtovt Tbit document eo nto in t__ IL- pages Tbit is copy ^ of tor lot tL Glassification Eoch perton who rtcoivot thltdocument must tipn the cover thstt I Route to Notsd by Date Route to Noted by D aft OHO A im *[|S 5(M) CRYSTAL STRUCTURES OF AMERICIUM COMPOUNDS1 D. H. Templeton and Carol H. Dauben Department of Chemistry and Radiation Laboratory University of California, Berkeley, California February 3, 1953 ABSTRACT The crystal structures of several compounds of americium, element 95, have been determined by the X-ray powder diffraction method. AisF^ is hexagonal (LaF^ type) with a = 4. 067 t 0.001 X and c « 7. 225 t 0.002 Si for the pseudoceil which explains the powder data. AmO^ is cubic (CaF^ type) with 1 a = 5. 38) t 0.001 X AmOCl is tetragonal (PbFCl type) with a = 4.00+O.OlX, c = 6.78 t 0.01 X. The metal parameter la 0.18 t 0.01. -

Nuclear Chemistry Card Sort
Nuclear Chemistry Card Sort Submitted by: Annette Gillespie, Ashely Gilomen, Sarah Johnson, and Stephanie Kimberlin Appalachian Regional Commission/Oak Ridge National Laboratory Summer Math-Science-Technology Institute Grade: 9-12 Lesson Duration: 45-60 minutes Materials Needed: • Media for Venn Diagram (e.g., poster board, white board, butcher paper, electronic devices) • One copy of the list of nuclear chemistry concepts/nuclear chemistry concept cards provided in this lesson plan • Envelope for storing cut-up list/cards Background Information: Students should be able to describe the basic structure of an atom and have a basic understanding of nuclear notation. Lesson Objective: Students will be able to • Describe the basic principles of nuclear chemistry. • Discern between diagrams and representations of nuclear equations and processes. • Use context clues to correctly categorize nuclear events. Instructional Process: All informative resources should be made available to the students so they can categorize the concepts (e.g., notes, text books, Internet). This lesson is best suited as an introduction activity for the beginning of the nuclear chemistry unit and/or as a review activity at the end of the nuclear chemistry unit. Students should be organized into groups of 3-4 to complete this activity. Each student group should create and label a three-circle Venn diagram on the teacher-designated media. (Refer to the examples provided in figure 1 and at the end of this lesson plan.) Each group should receive one copy of the list of nuclear chemistry concepts/nuclear chemistry concept cards, which they will cut up into individual cards. Once students have their diagrams drawn and cards cut out, they should place the nuclear chemistry concepts cards into one of three categories on the diagram: fission, fusion, or nuclear decay.