Distinct RNA N-Demethylation Pathways Catalyzed by Nonheme Iron ALKBH5 and FTO Enzymes Enable Regulation of Formaldehyde Release Rates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression
International Journal of Molecular Sciences Review Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression Guang Yang, Rachel Shi and Qing Zhang * Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; [email protected] (G.Y.); [email protected] (R.S.) * Correspondence: [email protected]; Tel.: +1-214-645-4671 Received: 6 October 2020; Accepted: 29 October 2020; Published: 31 October 2020 Abstract: Oxygen homeostasis regulation is the most fundamental cellular process for adjusting physiological oxygen variations, and its irregularity leads to various human diseases, including cancer. Hypoxia is closely associated with cancer development, and hypoxia/oxygen-sensing signaling plays critical roles in the modulation of cancer progression. The key molecules of the hypoxia/oxygen-sensing signaling include the transcriptional regulator hypoxia-inducible factor (HIF) which widely controls oxygen responsive genes, the central members of the 2-oxoglutarate (2-OG)-dependent dioxygenases, such as prolyl hydroxylase (PHD or EglN), and an E3 ubiquitin ligase component for HIF degeneration called von Hippel–Lindau (encoding protein pVHL). In this review, we summarize the current knowledge about the canonical hypoxia signaling, HIF transcription factors, and pVHL. In addition, the role of 2-OG-dependent enzymes, such as DNA/RNA-modifying enzymes, JmjC domain-containing enzymes, and prolyl hydroxylases, in gene regulation of cancer progression, is specifically reviewed. We also discuss the therapeutic advancement of targeting hypoxia and oxygen sensing pathways in cancer. Keywords: hypoxia; PHDs; TETs; JmjCs; HIFs 1. Introduction Molecular oxygen serves as a co-factor in many biochemical processes and is fundamental for aerobic organisms to maintain intracellular ATP levels [1,2]. -

Potential for and Distribution of Enzymatic Biodegradation of Polystyrene by Environmental Microorganisms
materials Communication Potential for and Distribution of Enzymatic Biodegradation of Polystyrene by Environmental Microorganisms Liyuan Hou and Erica L.-W. Majumder * Department of Chemistry, SUNY College of Environmental Science and Forestry, Syracuse, NY 13210, USA; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +1-3154706854 Abstract: Polystyrene (PS) is one of the main polymer types of plastic wastes and is known to be resistant to biodegradation, resulting in PS waste persistence in the environment. Although previous studies have reported that some microorganisms can degrade PS, enzymes and mechanisms of microorganism PS biodegradation are still unknown. In this study, we summarized microbial species that have been identified to degrade PS. By screening the available genome information of microorganisms that have been reported to degrade PS for enzymes with functional potential to depolymerize PS, we predicted target PS-degrading enzymes. We found that cytochrome P4500s, alkane hydroxylases and monooxygenases ranked as the top potential enzyme classes that can degrade PS since they can break C–C bonds. Ring-hydroxylating dioxygenases may be able to break the side-chain of PS and oxidize the aromatic ring compounds generated from the decomposition of PS. These target enzymes were distributed in Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes, suggesting a broad potential for PS biodegradation in various earth environments and microbiomes. Our results provide insight into the enzymatic degradation of PS and suggestions for realizing the biodegradation of this recalcitrant plastic. Citation: Hou, L.; Majumder, E.L. Keywords: plastics; polystyrene biodegradation; enzymatic biodegradation; monooxygenase; alkane Potential for and Distribution of hydroxylase; cytochrome P450 Enzymatic Biodegradation of Polystyrene by Environmental Microorganisms. -

Dn056490.Pdf
A Rapid Transcriptome Response Is Associated with Desiccation Resistance in Aerially-Exposed Killifish Embryos Ange`le Tingaud-Sequeira1¤a, Juan-Jose´ Lozano2, Cinta Zapater1, David Otero1, Michael Kube3¤b, Richard Reinhardt3¤c, Joan Cerda` 1* 1 Institut de Recerca i Tecnologia Agroalimenta`ries (IRTA)-Institut de Cie`ncies del Mar, CSIC, Barcelona, Spain, 2 Bioinformatics Platform, CIBERHED, Barcelona, Spain, 3 Max Planck Institute for Molecular Genetics, Berlin-Dahlem, Germany Abstract Delayed hatching is a form of dormancy evolved in some amphibian and fish embryos to cope with environmental conditions transiently hostile to the survival of hatchlings or larvae. While diapause and cryptobiosis have been extensively studied in several animals, very little is known concerning the molecular mechanisms involved in the sensing and response of fish embryos to environmental cues. Embryos of the euryhaline killifish Fundulus heteroclitus advance dvelopment when exposed to air but hatching is suspended until flooding with seawater. Here, we investigated how transcriptome regulation underpins this adaptive response by examining changes in gene expression profiles of aerially incubated killifish embryos at ,100% relative humidity, compared to embryos continuously flooded in water. The results confirm that mid-gastrula embryos are able to stimulate development in response to aerial incubation, which is accompanied by the differential expression of at least 806 distinct genes during a 24 h period. Most of these genes (,70%) appear to be differentially expressed within 3 h of aerial exposure, suggesting a broad and rapid transcriptomic response. This response seems to include an early sensing phase, which overlaps with a tissue remodeling and activation of embryonic development phase involving many regulatory and metabolic pathways. -

Human Alkb Homologue 5 Is a Nuclear 2-Oxoglutarate Dependent Oxygenase and a Direct Target of Hypoxia- Inducible Factor 1A (HIF-1A)
Human AlkB Homologue 5 Is a Nuclear 2-Oxoglutarate Dependent Oxygenase and a Direct Target of Hypoxia- Inducible Factor 1a (HIF-1a) Armin Thalhammer2, Zuzana Bencokova3., Rachel Poole3., Christoph Loenarz2., Julie Adam1, Linda O’Flaherty1, Johannes Scho¨ del1, David Mole1, Konstantinos Giaslakiotis4, Christopher J. Schofield2, Ester M. Hammond3, Peter J. Ratcliffe1, Patrick J. Pollard1* 1 Henry Wellcome Building for Molecular Physiology, University of Oxford, Oxford, United Kingdom, 2 Chemistry Research Laboratory and The Oxford Centre for Integrative Systems Biology, Department of Chemistry, University of Oxford, Oxford, United Kingdom, 3 Gray Institute for Radiation Oncology and Biology, University of Oxford, Oxford, United Kingdom, 4 Department of Cellular Pathology, John Radcliffe Hospital, Oxford, United Kingdom Abstract Human 2-oxoglutarate oxygenases catalyse a range of biological oxidations including the demethylation of histone and nucleic acid substrates and the hydroxylation of proteins and small molecules. Some of these processes are centrally involved in regulation of cellular responses to hypoxia. The ALKBH proteins are a sub-family of 2OG oxygenases that are defined by homology to the Escherichia coli DNA-methylation repair enzyme AlkB. Here we report evidence that ALKBH5 is probably unique amongst the ALKBH genes in being a direct transcriptional target of hypoxia inducible factor-1 (HIF-1) and is induced by hypoxia in a range of cell types. We show that purified recombinant ALKBH5 is a bona fide 2OG oxygenase that catalyses the decarboxylation of 2OG but appears to have different prime substrate requirements from those so far defined for other ALKBH family members. Our findings define a new class of HIF-transcriptional target gene and suggest that ALKBH5 may have a role in the regulation of cellular responses to hypoxia. -

Identifying Functional Roles for Alkb in the Adaptive
IDENTIFYING FUNCTIONAL ROLES FOR ALKB IN THE ADAPTIVE RESPONSE OF ESCHERICHIA COLI TO ALKYLATION DAMAGE SUNEET DINGLAY A thesis submitted for Ph.D.; 2000 Imperial Cancer Research Fund, Clare Hall Laboratories, Blanche Lane, South Mimms, Herts, EN6 3LD & University College London, Gower Street, London, WC1E 6BT ProQuest Number: 10608883 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10608883 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 In loving memory of my Papa ji. ABSTRACT In 1977 a novel, inducible and error free DNA repair system in Escherichia coli came to light. It protected E. coli against the mutagenic and cytotoxic effects of alkylating agents such as N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) and methyl methanesulfonate (MMS), and was termed the ‘adaptive response of E. coli to alkylation damage’. This response consists of four inducible genes; ada, aidB, alkA and alkB. The ada gene product encodes an 06-methylguanine- DNA methyltransferase, and is also the positive regulator of the response. The alkA gene product encodes a 3-methyladenine- DNA glycosylase, and aidB shows homology to several mammalian acyl coenzyme A dehydrogenases. -

Electronic Supplementary Material (ESI) for Metallomics
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2018 Uniprot Entry name Gene names Protein names Predicted Pattern Number of Iron role EC number Subcellular Membrane Involvement in disease Gene ontology (biological process) Id iron ions location associated 1 P46952 3HAO_HUMAN HAAO 3-hydroxyanthranilate 3,4- H47-E53-H91 1 Fe cation Catalytic 1.13.11.6 Cytoplasm No NAD biosynthetic process [GO:0009435]; neuron cellular homeostasis dioxygenase (EC 1.13.11.6) (3- [GO:0070050]; quinolinate biosynthetic process [GO:0019805]; response to hydroxyanthranilate oxygenase) cadmium ion [GO:0046686]; response to zinc ion [GO:0010043]; tryptophan (3-HAO) (3-hydroxyanthranilic catabolic process [GO:0006569] acid dioxygenase) (HAD) 2 O00767 ACOD_HUMAN SCD Acyl-CoA desaturase (EC H120-H125-H157-H161; 2 Fe cations Catalytic 1.14.19.1 Endoplasmic Yes long-chain fatty-acyl-CoA biosynthetic process [GO:0035338]; unsaturated fatty 1.14.19.1) (Delta(9)-desaturase) H160-H269-H298-H302 reticulum acid biosynthetic process [GO:0006636] (Delta-9 desaturase) (Fatty acid desaturase) (Stearoyl-CoA desaturase) (hSCD1) 3 Q6ZNF0 ACP7_HUMAN ACP7 PAPL PAPL1 Acid phosphatase type 7 (EC D141-D170-Y173-H335 1 Fe cation Catalytic 3.1.3.2 Extracellular No 3.1.3.2) (Purple acid space phosphatase long form) 4 Q96SZ5 AEDO_HUMAN ADO C10orf22 2-aminoethanethiol dioxygenase H112-H114-H193 1 Fe cation Catalytic 1.13.11.19 Unknown No oxidation-reduction process [GO:0055114]; sulfur amino acid catabolic process (EC 1.13.11.19) (Cysteamine -

Cyclooxygenase 2-Dependent and Independent Activation of Akt
Shimada et al. BMC Urology 2011, 11:8 http://www.biomedcentral.com/1471-2490/11/8 RESEARCHARTICLE Open Access Cyclooxygenase 2-dependent and independent activation of Akt through casein kinase 2a contributes to human bladder cancer cell survival Keiji Shimada1*, Satoshi Anai2, Develasco A Marco3, Kiyohide Fujimoto2 and Noboru Konishi1 Abstract Background: Survival rate for patients presenting muscle invasive bladder cancer is very low, and useful therapeutic target has not been identified yet. In the present study, new COX2 downstream signals involved in urothelial carcinoma cell survival were investigated in vitro and in vivo. Methods: COX2 gene was silenced by siRNA transfection. Orthotopic implantation animal model and transurethral instillation of siRNA with atelocollagen was constructed to examine the effects of COX2 knockdown in vivo. Cell cycle was examined by flowcytoketry. Surgical specimens derived from patients with urinary bladder cancer (all were initially diagnosed cases) were used for immunohistochemical analysis of the indicated protein expression in urothelial carcinoma cells. Results: Treatment with the COX2 inhibitor or knockdown of COX2 reduced expression of casein kinase (CK) 2 a,a phophorylated Akt and urokinase type plasminogen activator (uPA), resulting in p27 induction, cell cycle arrest at G1 phase and cell growth suppression in human urothelial carcinoma cell lines expressing COX2. Silencing of CK2a exhibited the similar effects. Even in UMUC3 cells lacking the COX2 gene, COX2 inhibition also inhibited cell growth through down-regulation of the CK2a-Akt/uPA axis. The mouse orthotropic bladder cancer model demonstrated that the COX2 inhibitor, meloxicam significantly reduced CK2a, phosphorylated Akt and uPA expression, whereas induced p27 by which growth and invasiveness of bladder cancer cells were strongly inhibited. -

Selection of Endophytic Strains for Enhanced Bacteria-Assisted Phytoremediation of Organic Pollutants Posing a Public Health Hazard
International Journal of Molecular Sciences Review Selection of Endophytic Strains for Enhanced Bacteria-Assisted Phytoremediation of Organic Pollutants Posing a Public Health Hazard Magdalena Anna Kara´s*, Sylwia Wdowiak-Wróbel and Wojciech Sokołowski Department of Genetics and Microbiology, Institute of Biological Sciences, Faculty of Biology and Biotechnology, Maria Curie-Skłodowska University, Akademicka 19, 20-033 Lublin, Poland; [email protected] (S.W.-W.); [email protected] (W.S.) * Correspondence: [email protected]; Tel.: +48-81-537-50-58 Abstract: Anthropogenic activities generate a high quantity of organic pollutants, which have an impact on human health and cause adverse environmental effects. Monitoring of many hazardous contaminations is subject to legal regulations, but some substances such as therapeutic agents, personal care products, hormones, and derivatives of common organic compounds are currently not included in these regulations. Classical methods of removal of organic pollutants involve economically challenging processes. In this regard, remediation with biological agents can be an alternative. For in situ decontamination, the plant-based approach called phytoremediation can be used. However, the main disadvantages of this method are the limited accumulation capacity of plants, sensitivity to the action of high concentrations of hazardous pollutants, and no possibility of using pollutants for growth. To overcome these drawbacks and additionally increase the efficiency of Citation: Kara´s,M.A.; the process, an integrated technology of bacteria-assisted phytoremediation is being used recently. Wdowiak-Wróbel, S.; Sokołowski, W. For the system to work, it is necessary to properly select partners, especially endophytes for specific Selection of Endophytic Strains for Enhanced Bacteria-Assisted plants, based on the knowledge of their metabolic abilities and plant colonization capacity. -

Table 4. 391 Probe Sets Still Rhythmic After Sleep Deprivation
Table 4. 391 probe sets still rhythmic after sleep deprivation Affymetrix ID Gene Symbol Description Accession time_sin time_cos adj.P.Val 1438211_s_at Dbp D site albumin promoter binding protein BB550183 -0.013 -0.877 1.44E-13 1418174_at Dbp D site albumin promoter binding protein BC018323 -0.036 -0.880 1.74E-13 1425099_a_at Arntl aryl hydrocarbon receptor nuclear translocator-like BC011080 0.135 0.418 1.02E-11 1416958_at Nr1d2 nuclear receptor subfamily 1, group D, member 2 NM_011584 -0.035 -0.397 7.53E-11 1421087_at Per3 period homolog 3 (Drosophila) NM_011067 -0.119 -0.477 3.46E-10 1450779_at Fabp7 fatty acid binding protein 7, brain NM_021272 0.433 0.404 3.82E-10 1424175_at Tef thyrotroph embryonic factor BC017689 -0.113 -0.279 1.39E-09 1435188_at Gm129 gene model 129, (NCBI) BB407125 -0.097 -0.667 4.17E-09 1417602_at Per2 period homolog 2 (Drosophila) AF035830 -0.460 -0.408 5.97E-09 1425560_a_at S100a16 S100 calcium binding protein A16 BC020031 0.228 0.194 5.97E-09 1435459_at Fmo2 flavin containing monooxygenase 2 BM936480 -0.255 -0.426 6.23E-09 1457350_at Per2 period homolog 2 (Drosophila) BG298986 -0.312 -0.485 7.81E-09 1445892_at Per2 Period homolog 2 (Drosophila) BM238318 -0.300 -0.432 1.50E-08 1448383_at Mmp14 matrix metallopeptidase 14 (membrane-inserted) NM_008608 0.082 0.474 3.74E-08 1456046_at Cd93 CD93 antigen AV319144 0.357 0.320 4.89E-08 1429286_at 1190003M12Rik RIKEN cDNA 1190003M12 gene AK004474 -0.511 -0.245 5.08E-08 similar to Putative RNA-binding protein 3 (RNA- 1422660_at LOC671237 AY052560 -0.269 -0.318 5.40E-08 -

Human Alkb Homologue 5 Is a Nuclear 2-Oxoglutarate Dependent Oxygenase and a Direct Target of Hypoxia-Inducible Factor 1 (HIF
Edinburgh Research Explorer Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1 (HIF- 1) Citation for published version: Thalhammer, A, Bencokova, Z, Poole, R, Loenarz, C, Adam, J, O'Flaherty, L, Schödel, J, Mole, D, Giaslakiotis, K, Schofield, CJ, Hammond, EM, Ratcliffe, PJ & Pollard, PJ 2011, 'Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1 (HIF-1)', PLoS ONE, vol. 6, no. 1, pp. e16210. https://doi.org/10.1371/journal.pone.0016210 Digital Object Identifier (DOI): 10.1371/journal.pone.0016210 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: PLoS ONE Publisher Rights Statement: Copyright: © 2011 Thallhammer et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. -

Combining Trna Sequencing Methods to Characterize Plant Trna Expression and Post- Transcriptional Modification
bioRxiv preprint doi: https://doi.org/10.1101/790451; this version posted October 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Combining tRNA sequencing methods to characterize plant tRNA expression and post- transcriptional modification Jessica M. Warren1, Thalia Salinas-Giegé2, Guillaume Hummel2, Nicole L. Coots1, Joshua M. Svendsen1, Kristen C. Brown1, Laurence Maréchal-Drouard2, Daniel B. Sloan1 1Department of Biology, Colorado State University, Fort Collins, CO 80523-1878, USA 2Institut de biologie moléculaire des plantes-CNRS, Université de Strasbourg, F-67084 Strasbourg, France Corresponding author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/790451; this version posted October 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. ABSTRACT Differences in tRNA expression have been implicated in a remarkable number of biological processes. There is growing evidence that tRNA genes can play dramatically different roles depending on both expression and post-transcriptional modification, yet sequencing tRNAs to measure abundance and detect modifications remains challenging. Their secondary structure and extensive post-transcriptional modifications interfere with RNA-seq library preparation methods and have limited the utility of high- throughput sequencing technologies. Here, we combine two modifications to standard RNA-seq methods by treating with the demethylating enzyme AlkB and ligating with tRNA-specific adapters in order to sequence tRNAs from four species of flowering plants, a group that has been shown to have some of the most extensive rates of post-transcriptional tRNA modifications. -
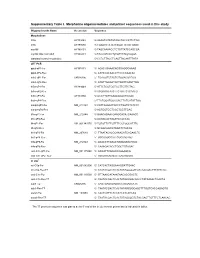
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC