Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Network Propagation Approach to Prioritize Long Tail Genes in Cancer
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.05.429983; this version posted February 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Network Propagation Approach to Prioritize Long Tail Genes in Cancer Hussein Mohsen1,*, Vignesh Gunasekharan2, Tao Qing2, Sahand Negahban3, Zoltan Szallasi4, Lajos Pusztai2,*, Mark B. Gerstein1,5,6,3,* 1 Computational Biology & Bioinformatics Program, Yale University, New Haven, CT 06511, USA 2 Breast Medical Oncology, Yale School of Medicine, New Haven, CT 06511, USA 3 Department of Statistics & Data Science, Yale University, New Haven, CT 06511, USA 4 Children’s Hospital Informatics Program, Harvard-MIT Division of Health Sciences and Technology, Harvard Medical School, Boston, MA 02115, USA 5 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA 6 Department of Computer Science, Yale University, New Haven, CT 06511, USA * Corresponding author Abstract Introduction. The diversity of genomic alterations in cancer pose challenges to fully understanding the etiologies of the disease. Recent interest in infrequent mutations, in genes that reside in the “long tail” of the mutational distribution, uncovered new genes with significant implication in cancer development. The study of these genes often requires integrative approaches with multiple types of biological data. Network propagation methods have demonstrated high efficacy in uncovering genomic patterns underlying cancer using biological interaction networks. Yet, the majority of these analyses have focused their assessment on detecting known cancer genes or identifying altered subnetworks. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

SLFN11 Promotes CDT1 Degradation by CUL4 in Response to Replicative DNA Damage, While Its Absence Leads to Synthetic Lethality with ATR/CHK1 Inhibitors
SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors Ukhyun Joa,1, Yasuhisa Muraia, Sirisha Chakkab, Lu Chenb, Ken Chengb, Junko Muraic, Liton Kumar Sahaa, Lisa M. Miller Jenkinsd, and Yves Pommiera,1 aDevelopmental Therapeutics Branch, Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20814; bNational Center for Advancing Translational Sciences, Functional Genomics Laboratory, NIH, Rockville, MD 20850; cInstitute for Advanced Biosciences, Keio University, 997-0052 Yamagata, Japan; and dLaboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD 20892 Edited by Richard D. Kolodner, Ludwig Institute for Cancer Research, La Jolla, CA, and approved December 8, 2020 (received for review July 29, 2020) Schlafen-11 (SLFN11) inactivation in ∼50% of cancer cells confers condensation related to deposition of H3K27me3 in the gene broad chemoresistance. To identify therapeutic targets and under- body of SLFN11 by EZH2, a histone methyltransferase (11). lying molecular mechanisms for overcoming chemoresistance, we Targeting epigenetic regulators is therefore an attractive com- performed an unbiased genome-wide RNAi screen in SLFN11-WT bination strategy to overcome chemoresistance of SLFN11- and -knockout (KO) cells. We found that inactivation of Ataxia deficient cancers (10, 25, 26). An alternative approach is to at- Telangiectasia- and Rad3-related (ATR), CHK1, BRCA2, and RPA1 tack SLFN11-negative cancer cells by targeting the essential SLFN11 overcome chemoresistance to camptothecin (CPT) in -KO pathways that cells use to overcome replicative damage and cells. Accordingly, we validate that clinical inhibitors of ATR replication stress. -

Download Author Version (PDF)
Molecular BioSystems Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/molecularbiosystems Page 1 of 29 Molecular BioSystems Mutated Genes and Driver Pathways Involved in Myelodysplastic Syndromes—A Transcriptome Sequencing Based Approach Liang Liu1*, Hongyan Wang1*, Jianguo Wen2*, Chih-En Tseng2,3*, Youli Zu2, Chung-che Chang4§, Xiaobo Zhou1§ 1 Center for Bioinformatics and Systems Biology, Division of Radiologic Sciences, Wake Forest University Baptist Medical Center, Winston-Salem, NC 27157, USA. 2 Department of Pathology, the Methodist Hospital Research Institute, -

Phosphoproteomics of Retinoblastoma: a Pilot Study Identifies Aberrant Kinases
molecules Article Phosphoproteomics of Retinoblastoma: A Pilot Study Identifies Aberrant Kinases Lakshmi Dhevi Nagarajha Selvan 1,†, Ravikanth Danda 1,2,†, Anil K. Madugundu 3 ID , Vinuth N. Puttamallesh 3, Gajanan J. Sathe 3,4, Uma Maheswari Krishnan 2, Vikas Khetan 5, Pukhraj Rishi 5, Thottethodi Subrahmanya Keshava Prasad 3,6 ID , Akhilesh Pandey 3,7,8, Subramanian Krishnakumar 1, Harsha Gowda 3,* and Sailaja V. Elchuri 9,* 1 L&T Opthalmic Pathology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (L.D.N.S.); [email protected] (R.D.); [email protected] (S.K.) 2 Centre for Nanotechnology and Advanced Biomaterials, Shanmugha Arts, Science, Technology and Research Academy University, Tanjore, Tamil Nadu 613 401, India; [email protected] 3 Institute of Bioinformatics, International Technology Park, Bangalore, Karnataka 560 066, India; [email protected] (A.K.M.); [email protected] (V.N.P.); [email protected] (G.J.S.); [email protected] (T.S.K.P.); [email protected] (A.P.) 4 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576 104, India 5 Shri Bhagwan Mahavir Vitreoretinal Services, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (V.K.); [email protected] (P.R.) 6 Center for Systems Biology and Molecular Medicine, Yenepoya Research Centre, Yenepoya (Deemed to be University), Mangalore, Karnataka 575 108, India 7 McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 8 Departments of Biological Chemistry, Pathology and Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 9 Department of Nanotechnology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India * Correspondence: [email protected] (H.G.); [email protected] (S.V.E.); Tel.: +91-80-28416140 (H.G.); +91-44-28271616 (S.V.E.) † These authors contributed equally to this work. -
Attachment PDF Icon
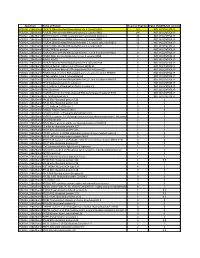
Spectrum Name of Protein Count of Peptides Ratio (POL2RA/IgG control) POLR2A_228kdBand POLR2A DNA-directed RNA polymerase II subunit RPB1 197 NOT IN CONTROL IP POLR2A_228kdBand POLR2B DNA-directed RNA polymerase II subunit RPB2 146 NOT IN CONTROL IP POLR2A_228kdBand RPAP2 Isoform 1 of RNA polymerase II-associated protein 2 24 NOT IN CONTROL IP POLR2A_228kdBand POLR2G DNA-directed RNA polymerase II subunit RPB7 23 NOT IN CONTROL IP POLR2A_228kdBand POLR2H DNA-directed RNA polymerases I, II, and III subunit RPABC3 19 NOT IN CONTROL IP POLR2A_228kdBand POLR2C DNA-directed RNA polymerase II subunit RPB3 17 NOT IN CONTROL IP POLR2A_228kdBand POLR2J RPB11a protein 7 NOT IN CONTROL IP POLR2A_228kdBand POLR2E DNA-directed RNA polymerases I, II, and III subunit RPABC1 8 NOT IN CONTROL IP POLR2A_228kdBand POLR2I DNA-directed RNA polymerase II subunit RPB9 9 NOT IN CONTROL IP POLR2A_228kdBand ALMS1 ALMS1 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2D DNA-directed RNA polymerase II subunit RPB4 6 NOT IN CONTROL IP POLR2A_228kdBand GRINL1A;Gcom1 Isoform 12 of Protein GRINL1A 6 NOT IN CONTROL IP POLR2A_228kdBand RECQL5 Isoform Beta of ATP-dependent DNA helicase Q5 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2L DNA-directed RNA polymerases I, II, and III subunit RPABC5 5 NOT IN CONTROL IP POLR2A_228kdBand KRT6A Keratin, type II cytoskeletal 6A 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2K DNA-directed RNA polymerases I, II, and III subunit RPABC4 2 NOT IN CONTROL IP POLR2A_228kdBand RFC4 Replication factor C subunit 4 1 NOT IN CONTROL IP POLR2A_228kdBand RFC2 -

MAF1 Represses CDKN1A Through a Pol III-Dependent Mechanism Yu-Ling Lee, Yuan-Ching Li, Chia-Hsin Su, Chun-Hui Chiao, I-Hsuan Lin, Ming-Ta Hsu*
RESEARCH ARTICLE elifesciences.org MAF1 represses CDKN1A through a Pol III-dependent mechanism Yu-Ling Lee, Yuan-Ching Li, Chia-Hsin Su, Chun-Hui Chiao, I-Hsuan Lin, Ming-Ta Hsu* Institute of Biochemistry and Molecular Biology, School of Life Science, National Yang-Ming University, Taipei, Taiwan Abstract MAF1 represses Pol III-mediated transcription by interfering with TFIIIB and Pol III. Herein, we found that MAF1 knockdown induced CDKN1A transcription and chromatin looping concurrently with Pol III recruitment. Simultaneous knockdown of MAF1 with Pol III or BRF1 (subunit of TFIIIB) diminished the activation and looping effect, which indicates that recruiting Pol III was required for activation of Pol II-mediated transcription and chromatin looping. Chromatin- immunoprecipitation analysis after MAF1 knockdown indicated enhanced binding of Pol III and BRF1, as well as of CFP1, p300, and PCAF, which are factors that mediate active histone marks, along with the binding of TATA binding protein (TBP) and POLR2E to the CDKN1A promoter. Simultaneous knockdown with Pol III abolished these regulatory events. Similar results were obtained for GDF15. Our results reveal a novel mechanism by which MAF1 and Pol III regulate the activity of a protein- coding gene transcribed by Pol II. DOI: 10.7554/eLife.06283.001 Introduction Transcription by RNA polymerase III (Pol III) is regulated by MAF1, which is a highly conserved protein in eukaryotes (Pluta et al., 2001; Reina et al., 2006). MAF1 represses Pol III transcription through *For correspondence: mth@ym. association with BRF1, a subunit of initiation factor TFIIIB, which prevents attachment of TFIIIB onto edu.tw DNA. -

The Human Genome Project
TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 Contents FOREWORD . 2 THE GENOME PROJECT—WHY THE DOE? . 4 A bold but logical step INTRODUCING THE HUMAN GENOME . 6 The recipe for life Some definitions . 6 A plan of action . 8 EXPLORING THE GENOMIC LANDSCAPE . 10 Mapping the terrain Two giant steps: Chromosomes 16 and 19 . 12 Getting down to details: Sequencing the genome . 16 Shotguns and transposons . 20 How good is good enough? . 26 Sidebar: Tools of the Trade . 17 Sidebar: The Mighty Mouse . 24 BEYOND BIOLOGY . 27 Instrumentation and informatics Smaller is better—And other developments . 27 Dealing with the data . 30 ETHICAL, LEGAL, AND SOCIAL IMPLICATIONS . 32 An essential dimension of genome research Foreword T THE END OF THE ROAD in Little has been rapid, and it is now generally agreed Cottonwood Canyon, near Salt that this international project will produce Lake City, Alta is a place of the complete sequence of the human genome near-mythic renown among by the year 2005. A skiers. In time it may well And what is more important, the value assume similar status among molecular of the project also appears beyond doubt. geneticists. In December 1984, a conference Genome research is revolutionizing biology there, co-sponsored by the U.S. Department and biotechnology, and providing a vital of Energy, pondered a single question: Does thrust to the increasingly broad scope of the modern DNA research offer a way of detect- biological sciences. -

Application of Microrna Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease
Article Application of microRNA Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease Jennifer L. Major, Rushita A. Bagchi * and Julie Pires da Silva * Department of Medicine, Division of Cardiology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; [email protected] * Correspondence: [email protected] (R.A.B.); [email protected] (J.P.d.S.) Supplementary Tables Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 www.mdpi.com/journal/mps Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 2 of 25 Table 1. List of all hsa-miRs identified by Human microRNA Disease Database (HMDD; v3.2) analysis. hsa-miRs were identified using the term “genetics” and “circulating” as input in HMDD. Targets CAD hsa-miR-1 Targets IR injury hsa-miR-423 Targets Obesity hsa-miR-499 hsa-miR-146a Circulating Obesity Genetics CAD hsa-miR-423 hsa-miR-146a Circulating CAD hsa-miR-149 hsa-miR-499 Circulating IR Injury hsa-miR-146a Circulating Obesity hsa-miR-122 Genetics Stroke Circulating CAD hsa-miR-122 Circulating Stroke hsa-miR-122 Genetics Obesity Circulating Stroke hsa-miR-26b hsa-miR-17 hsa-miR-223 Targets CAD hsa-miR-340 hsa-miR-34a hsa-miR-92a hsa-miR-126 Circulating Obesity Targets IR injury hsa-miR-21 hsa-miR-423 hsa-miR-126 hsa-miR-143 Targets Obesity hsa-miR-21 hsa-miR-223 hsa-miR-34a hsa-miR-17 Targets CAD hsa-miR-223 hsa-miR-92a hsa-miR-126 Targets IR injury hsa-miR-155 hsa-miR-21 Circulating CAD hsa-miR-126 hsa-miR-145 hsa-miR-21 Targets Obesity hsa-mir-223 hsa-mir-499 hsa-mir-574 Targets IR injury hsa-mir-21 Circulating IR injury Targets Obesity hsa-mir-21 Targets CAD hsa-mir-22 hsa-mir-133a Targets IR injury hsa-mir-155 hsa-mir-21 Circulating Stroke hsa-mir-145 hsa-mir-146b Targets Obesity hsa-mir-21 hsa-mir-29b Methods Protoc. -

Human Prefoldin Modulates Co-Transcriptional Pre-Mrna Splicing
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.14.150466; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. BIOLOGICAL SCIENCES: Biochemistry Human prefoldin modulates co-transcriptional pre-mRNA splicing Payán-Bravo L 1,2, Peñate X 1,2 *, Cases I 3, Pareja-Sánchez Y 1, Fontalva S 1,2, Odriozola Y 1,2, Lara E 1, Jimeno-González S 2,5, Suñé C 4, Reyes JC 5, Chávez S 1,2. 1 Instituto de Biomedicina de Sevilla, Universidad de Sevilla-CSIC-Hospital Universitario V. del Rocío, Seville, Spain. 2 Departamento de Genética, Facultad de Biología, Universidad de Sevilla, Seville, Spain. 3 Centro Andaluz de Biología del Desarrollo, CSIC-Universidad Pablo de Olavide, Seville, Spain. 4 Department of Molecular Biology, Institute of Parasitology and Biomedicine "López Neyra" IPBLN-CSIC, PTS, Granada, Spain. 5 Andalusian Center of Molecular Biology and Regenerative Medicine-CABIMER, Junta de Andalucia-University of Pablo de Olavide-University of Seville-CSIC, Seville, Spain. Correspondence: Sebastián Chávez, IBiS, campus HUVR, Avda. Manuel Siurot s/n, Sevilla, 41013, Spain. Phone: +34-955923127: e-mail: [email protected]. * Co- corresponding; [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2020.06.14.150466; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Prefoldin is a heterohexameric complex conserved from archaea to humans that plays a cochaperone role during the cotranslational folding of actin and tubulin monomers. -

Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features
cancers Review Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features Valeria Visconte 1, Megan O. Nakashima 2 and Heesun J. Rogers 2,* 1 Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] 2 Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-445-2719 Received: 15 October 2019; Accepted: 19 November 2019; Published: 22 November 2019 Abstract: Components of the pre-messenger RNA splicing machinery are frequently mutated in myeloid malignancies. Mutations in LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2 genes occur at various frequencies ranging between 40% and 85% in different subtypes of myelodysplastic syndrome (MDS) and 5% and 10% of acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs). In some instances, splicing factor (SF) mutations have provided diagnostic utility and information on clinical outcomes as exemplified by SF3B1 mutations associated with increased ring sideroblasts (RS) in MDS-RS or MDS/MPN-RS with thrombocytosis. SF3B1 mutations are associated with better survival outcomes, while SRSF2 mutations are associated with a shorter survival time and increased AML progression, and U2AF1 mutations with a lower remission rate and shorter survival time. Beside the presence of mutations, transcriptomics technologies have shown that one third of genes in AML patients are differentially expressed, leading to altered transcript stability, interruption of protein function, and improper translation compared to those of healthy individuals. The detection of SF mutations demonstrates the importance of splicing abnormalities in the hematopoiesis of MDS and AML patients given the fact that abnormal splicing regulates the function of several transcriptional factors (PU.1, RUNX1, etc.) crucial in hematopoietic function. -

A High Throughput, Functional Screen of Human Body Mass Index GWAS Loci Using Tissue-Specific Rnai Drosophila Melanogaster Crosses Thomas J
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. Baranski Washington University School of Medicine in St. Louis Aldi T. Kraja Washington University School of Medicine in St. Louis Jill L. Fink Washington University School of Medicine in St. Louis Mary Feitosa Washington University School of Medicine in St. Louis Petra A. Lenzini Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Baranski, Thomas J.; Kraja, Aldi T.; Fink, Jill L.; Feitosa, Mary; Lenzini, Petra A.; Borecki, Ingrid B.; Liu, Ching-Ti; Cupples, L. Adrienne; North, Kari E.; and Province, Michael A., ,"A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses." PLoS Genetics.14,4. e1007222. (2018). https://digitalcommons.wustl.edu/open_access_pubs/6820 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Thomas J. Baranski, Aldi T. Kraja, Jill L. Fink, Mary Feitosa, Petra A. Lenzini, Ingrid B. Borecki, Ching-Ti Liu, L. Adrienne Cupples, Kari E. North, and Michael A. Province This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6820 RESEARCH ARTICLE A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J.