Clathrin-Independent Pathways Do Not Contribute Significantly to Endocytic 2 Flux
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Palmitoylation of Caveolin-1 and Its Importance for Structural and Functional Plasticity
Palmitoylation of Caveolin-1 and its importance for structural and functional plasticity A DISSERTATION SUBMITTED TO THE FACULTY OF UNIVERSITY OF MINNESOTA BY Katherine R. Tonn IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Paul G. Mermelstein November 2018 © Katherine R. Tonn Eisinger 2018 Acknowledgments I would like to thank my adviser, Dr. Paul Mermelstein, for his mentorship, training, support, and scientific flexibility. Infinite thanks to past and present members of the Mermelstein and Meisel Labs, especially Dr. Brittni Peterson, Dr. Kelsey Moore, Dr. Laura Been, Dr. Luis Martinez, Dr. Valerie Hedges, and Dr. John Meitzen, for being a source of scientific inspiration and friendship. I could not have asked for a better friend and lab colleague than Dr. Kellie Gross, whose input has kept me sane and made me a better scientist. Thanks also to stellar undergrads Kerry Trotter, Julia Dworsky, and Sam Swanson for their help and energy. Thanks to the many people who collaborated or helped in some way on the experiments presented in this thesis, including Dr. Mark Thomas, Dr. Lorene Lanier, Dr. Mark Dell’Acqua, Dr. Kevin Woolfrey, Dr. Brian Head, Dr. Jing Tong, and Dr. John Meitzen. In particular, I am extremely grateful for the collaboration of Dr. Lorene Lanier, whose enthusiasm helped reinvigorate me and this project. Finally, thanks to the members of my thesis committee, Drs. Harry Orr, Timothy Ebner, Anna Lee, and Robert Meisel, for their encouragement and guidance. i Dedication For Mom, my first and most important example of what it is to be smart, versatile, and kind. -

Novel Missense Mutation in the Caveolin-3 Gene in a Belgian Family
1349 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2003.028217 on 16 August 2004. Downloaded from SHORT REPORT Novel missense mutation in the caveolin-3 gene in a Belgian family with rippling muscle disease P Y K Van den Bergh, J M Ge´rard, J A Elosegi, M U Manto, C Kubisch, B G H Schoser ............................................................................................................................... J Neurol Neurosurg Psychiatry 2004;75:1349–1351. doi: 10.1136/jnnp.2003.028217 too small’’). The patient was diagnosed as having fibromyal- Rippling muscle disease (RMD) is a rare muscle disorder gia, which led to secondary depression and loss of her job. characterised by muscle stiffness, exercise induced myalgia, Muscle weakness and pigmenturia were absent. The medical and cramp-like sensations. It is genetically heterogeneous history was remarkable for mild hypothyroidism, peptic and can be acquired, but most cases show autosomal oesophagitis, and tobacco related asthmatiform bronchitis dominant inheritance due to mutations in the caveolin-3 with emphysema. At age 38, she had a neurological work-up. (CAV3) gene. We report a novel heterozygous missense The cranial nerves, muscle bulk, muscle strength, and deep mutation in CAV3 in a Belgian family with autosomal tendon reflexes were normal, and plantar responses were dominant RMD. flexor. Tapping with the reflex hammer or with the finger on A 40 year old woman complained of fatigue, exercise the muscles provoked an immediate, short lasting, forceful induced muscle pain, and muscle cramps since the age of 35. contraction and/or painful local mounding lasting for several Neurological examination revealed percussion induced seconds. PIRCs were most pronounced in the sternocleido- rapid muscle contractions (PIRCs) and localised muscle mastoid, deltoid, biceps, brachioradialis, finger and wrist mounding on percussion; muscle rippling was not observed. -
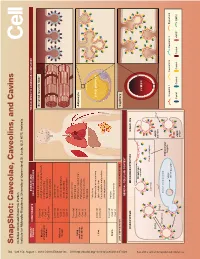
Snapshot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G
704 Cell SnapShot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G. Parton 154 Institute for Molecular Bioscience, University of Queensland, St. Lucia, QLD 4072, Australia , August 1,2013©2013 Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.07.009 ORGAN/ COMPONENTS DISEASE AND TISSUE-SPECIFIC CAVEOLAR COMPLEXES TISSUE CELLULAR PROCESS Rippling muscle disease Caveolin3 Striated muscle fiber Limb girdle muscular dystrophy Muscle Cavin1 skeletal and Cardiomyopathy Cavin4 cardiac Mechanoprotection Cav1 (heart) T-tubule formation Caveolin1 Lipodystrophy Caveolin2 Lipotoxicity Adipose Cavin1 Fatty acid regulation tissue Cavin2 Insulin signaling Cavin3 Mechanoprotection Atherosclerosis Caveolin1 Inammation Caveolin2 Lung Pulmonary hypertension and other Cavin1 Adipocyte Pulmonary brosis endothelia Cavin2 Mechanosensation Cavin3 Signaling Fatty liver Hepatocellular carcinoma Caveolin1 LIPID DROPLET Liver Lipid metabolism Caveolin2 Carbohydrate metabolism Liver regeneration Caveolin1 Autism Brain Cavin1 Schizophrenia Cavin3 LOW EXPRESSION HIGH EXPRESSION Capillary GENERAL CELLULAR CONTEXT ENDOCYTOSIS MECHANOPROTECTION SIGNALING See online version for legend and references. and legend for version online See Membrane stretch LUMEN Cavin complex Caveola EHD2 eNOS Ca2+ inactive Rab5 Dynamin2 EARLY ENDOSOME Intracellular targets MVB/ Caveolin 1 Caveolin 2 Caveolin 3 Dynamin2 late endosome eNOS Cavin1 Cavin2 Cavin3 Cavin4 eNOS EHD2 active SnapShot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G. Parton Institute for Molecular Bioscience, University of Queensland, St. Lucia, QLD 4072, Australia Caveolae, submicroscopic bulb-shaped plasma membrane pits, are an abundant feature of many mammalian cells (Parton and del Pozo, 2013). Caveolae and the major proteins of caveolae, caveolins (Rothberg et al., 1992), and cavins (Hill et al., 2008), are linked to a number of human diseases such as muscular dystrophy, cardiomyopathy, and lipodys- trophy (see Glossary) (Bruno et al., 1993; Fernández et al., 2006; Hayashi et al., 2009). -

Comprehensive Analysis of Lncrna‑Associated Cerna Network Reveals the Novel Potential of Lncrna, Mirna and Mrna Biomarkers in Human Rectosigmoid Junction Cancer
ONCOLOGY LETTERS 21: 144, 2021 Comprehensive analysis of lncRNA‑associated ceRNA network reveals the novel potential of lncRNA, miRNA and mRNA biomarkers in human rectosigmoid junction cancer QIANSHI ZHANG1*, ZHEN FENG1*, SHASHA SHI2, YU ZHANG3 and SHUANGYI REN1 Departments of 1Gastrointestinal Surgery, 2Ultrasound and 3Clinical Laboratory, The Second Affiliated Hospital of Dalian Medical University, Dalian, Liaoning 116023, P.R. China Received March 11, 2020; Accepted November 20, 2020 DOI: 10.3892/ol.2020.12405 Abstract. Although accumulating evidence has confirmed gene 20) and three mRNAs (sodium‑ and chloride‑dependent the potential biological functions of long non‑coding RNAs taurine transporter, fibroblast growth factor 13 and tubulin (lncRNAs) as competitive endogenous RNAs (ceRNAs) in polyglutamylase TTLL7) were significantly associated with colorectal tumorigenesis and progression, few studies have OS (P<0.05). Additionally, two lncRNAs (KCNQ1OT1 focused on rectosigmoid junction cancer. In the present and MIR17HG) interacted with most of the DEmiRNAs. study, a comprehensive analysis was conducted to explore Notably, two top‑ranked miRNAs (hsa‑miR‑374a‑5p and lncRNA‑mediated ceRNA implications and their potential hsa‑miR‑374b‑5p) associated networks were identified to be value for prognosis. lncRNA, microRNA (miR/miRNA) markedly associated with the pathogenesis. Furthermore, four and mRNA expression profiles were downloaded from DEmRNAs (caveolin‑1, MET, filamin‑A and AKT3) were The Cancer Genome Atlas database. Subsequently, a enriched in the Kyoto Encylopedia of Gene and Genomes lncRNA‑miRNA‑mRNA regulatory network was constructed pathway analysis, as well as being included in the ceRNA to evaluate the functions of these differentially expressed genes network. In summary, the present results revealed that a on overall survival (OS) for rectosigmoid junction cancer. -
Modulation of the Caveolin-3 Localization to Caveolae and STAT3 to Mitochondria by Catecholamine-Induced Cardiac Hypertrophy in H9c2 Cardiomyoblasts
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 41, No. 4, 226-235, April 2009 Modulation of the caveolin-3 localization to caveolae and STAT3 to mitochondria by catecholamine-induced cardiac hypertrophy in H9c2 cardiomyoblasts Kyuho Jeong*, Hayeong Kwon*, amine-induced cardiac hypertrophy. Chanhee Min and Yunbae Pak1 Keywords: cardiomegaly; caveolae; caveolin-3; cell Department of Biochemistry nucleus; heart; isoproterenol; mitochondria; phenyl- Division of Applied Life Science (BK21), PMBBRC ephrine; STAT3 transcription factor Environmental Biotechnology National Core Research Center Gyeongsang National University Jinju 660-701, Korea Introduction 1Corresponding author: Tel, 82-55-751-5961; Fax, 82-55-759-9363; E-mail, [email protected] Hypertension is major risk factors for cardiac da- mage, ischemia, myocardial infarction, and conge- *These authors contributed equally to this work. stive heart failure (Zampaglione et al., 1996). In DOI 10.3858/emm.2009.41.4.025 response to increased demands for cardiac work caused by various pathologic stresses, heart adapts Accepted 20 November 2008 through compensatory hypertrophy of myocytes. Abbreviations: Akt, protein kinase B; CsA, cyclosporin A; GPCR, Thus, cardiac hypertrophy is recognized in many G protein-coupled receptor; ISO, isoproterenol; PE, phenylephrine; cardiovascular diseases, such as hypertension, RTK, receptor tyrosine kinase; STAT3, signal transducers and acti- vascular disease, and myocardial infarction, and is vator of transcription 3 an independent risk factor for cardiac morbidity and mortality. Hypertrophic stimuli induce an in- crease in cell size in the absence of cell division through Ca2+ signaling and activation of PKC, Abstract MAPK and PKB/ Akt (Watanabe et al., 2001; Dorn and Force, 2005), and are accompanied by We investigated the effect of phenylephrine (PE)- and increased protein synthesis with reprogramming of isoproterenol (ISO)-induced cardiac hypertrophy on gene expression (Takeo et al., 2000). -

Caveolin-1 Expression in Schwann Cells
GLIA 27:39–52 (1999) Caveolin-1 Expression in Schwann Cells DANIEL D. MIKOL,* HOYLOND L. HONG, HSIN-LIN CHENG, AND EVA L. FELDMAN Department of Neurology, University of Michigan, Ann Arbor, Michigan KEY WORDS caveolae; cholesterol; differentiation; myelin; nerve ABSTRACT Caveolae are non-clathrin-coated invaginations of the plasma mem- brane, which are present in most cell types. An integral component of caveolae is the caveolin family of related proteins, which not only forms the structural framework of caveolae, but also likely subserves its functional roles, including regulation of signal transduction and cellular transport, in particular, cholesterol trafficking. Although caveolae have been identified ultrastructurally in the peripheral nervous system (PNS), caveolin expression has not previously been studied. To date, three caveolin genes have been reported. Here, we show for the first time that caveolin-1 is expressed by Schwann cells (SC) as well as several SC-derived cell lines. Caveolin-1 is enriched in the buoyant, detergent-insoluble membranes of rat sciatic nerve (SN) and SC, a hallmark of the caveolar compartment. Caveolin-1 exists as both soluble and insoluble forms in rat SN and SC, and localizes to SC cytoplasm and abaxonal myelin. SC caveolin-1 decreases after axotomy, when SC revert to a premyelinating phenotype. We speculate that caveolin-1 may regulate signal transduction and/or cholesterol transport in myelinating SC. GLIA 27:39–52, 1999. 1999 Wiley-Liss, Inc. INTRODUCTION A number of functions are proposed for caveolae: (1) the uptake of small molecules such as folate into cells, a Caveolae are plasma membrane microdomains that process known as potocytosis (Anderson et al., 1992); can invaginate to form 50–100 nm vesicles, and are (2) the transcytosis of molecules such as low density enriched in cholesterol, glycosphingolipids, a variety of lipoprotein across cells (Simionescu, 1983); (3) polar- signaling molecules, and the caveolins, a family of ized trafficking of proteins, especially in epithelial cells 18–24 kD proteins. -

Reproductionresearch
REPRODUCTIONRESEARCH The association between CDC42 and caveolin-1 is involved in the regulation of capacitation and acrosome reaction of guinea pig and mouse sperm R Baltie´rrez-Hoyos, A L Roa-Espitia and E O Herna´ndez-Gonza´lez Departamento de Biologı´a Celular, Centro de Investigacio´n y Estudios Avanzados del Instituto Polite´cnico Nacional, Avenida Instituto Polite´cnico Nacional 2508, San Pedro Zacatenco, Me´xico DF 07360, Mexico Correspondence should be addressed to E O Herna´ndez-Gonza´lez; Email: [email protected] Abstract In the mammalian sperm, the acrosome reaction (AR) is considered to be a regulated secretion that is an essential requirement for physiological fertilization. The AR is the all-or-nothing secretion system that allows for multiple membrane fusion events. It is a C Ca2 -regulated exocytosis reaction that has also been shown to be regulated by several signaling pathways. CDC42 has a central role in the regulated exocytosis through the activation of SNARE proteins and actin polymerization. Furthermore, the lipid raft protein caveolin- 1 (CAV1) functions as a scaffold and guanine nucleotide dissociation inhibitor protein for CDC42, which is inactivated when associated with CAV1. CDC42 and other RHO proteins have been shown to localize in the acrosome region of mammalian sperm; however, their relationship with the AR is unknown. Here, we present the first evidence that CDC42 and CAV1 could be involved in the regulation of capacitation and the AR. Our findings show that CDC42 is activated early during capacitation, reaching an activation maximum after 20 min of capacitation. Spontaneous and progesterone-induced ARs were inhibited when sperm were capacitated in presence of secramine A, a specific CDC42 inhibitor. -

Internalization of the TGF-Β Type I Receptor Into Caveolin-1 and EEA1 Double-Positive Early Endosomes
Cell Research (2015) 25:738-752. © 2015 IBCB, SIBS, CAS All rights reserved 1001-0602/15 $ 32.00 ORIGINAL ARTICLE www.nature.com/cr Internalization of the TGF-β type I receptor into caveolin-1 and EEA1 double-positive early endosomes Kangmin He1, 2, Xiaohua Yan3, Nan Li1, Song Dang1, Li Xu1, Bing Zhao3, Zijian Li2, Zhizhen Lv2, Xiaohong Fang1, Youyi Zhang2, Ye-Guang Chen3 1Beijing National Laboratory for Molecular Sciences, Key Laboratory of Molecular Nanostructures and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; 2Institute of Vascular Medicine Peking University Third Hos- pital and Academy for Advanced Interdisciplinary Studies, Peking University, Key Laboratory of Cardiovascular Molecular Biolo- gy and Regulatory Peptides, Ministry of Health, Key Laboratory of Molecular Cardiovascular Sciences, Ministry of Education and Beijing Key Laboratory of Cardiovascular Receptors Research, Beijing 100191,China; 3State Key Laboratory of Biomembrane and Membrane Biotechnology, Tsinghua-Peking Center for Life Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, China Endocytosis and intracellular sorting of transforming growth factor-β (TGF-β) receptors play an important reg- ulatory role in TGF-β signaling. Two major endocytic pathways, clathrin- and caveolae-mediated endocytosis, have been reported to independently mediate the internalization of TGF-β receptors. In this study, we demonstrate that the clathrin- and caveolae-mediated endocytic pathways can converge during TGF-β receptor endocytic trafficking. By tracking the intracellular dynamics of fluorescently-labeled TGF-β type I receptor (TβRI), we found that after me- diating TβRI internalization, certain clathrin-coated vesicles and caveolar vesicles are fused underneath the plasma membrane, forming a novel type of caveolin-1 and clathrin double-positive vesicles. -

Endocytosis: the Nanoparticle and Submicron Nanocompounds Gateway Into the Cell
pharmaceutics Review Endocytosis: The Nanoparticle and Submicron Nanocompounds Gateway into the Cell Darío Manzanares 1,2 and Valentín Ceña 1,2,* 1 Unidad Asociada Neurodeath, Universidad de Castilla-La Mancha, 02006 Albacete, Spain; [email protected] 2 CIBERNED, Instituto de Salud Carlos III, 28031 Madrid, Spain * Correspondence: [email protected] Received: 23 March 2020; Accepted: 15 April 2020; Published: 17 April 2020 Abstract: Nanoparticles (NPs) and submicron particles are increasingly used as carriers for delivering therapeutic compounds to cells. Their entry into the cell represents the initial step in this delivery process, being most of the nanoparticles taken up by endocytosis, although other mechanisms can contribute to the uptake. To increase the delivery efficiency of therapeutic compounds by NPs and submicron particles is very relevant to understand the mechanisms involved in the uptake process. This review covers the proposed pathways involved in the cellular uptake of different NPs and submicron particles types as well as the role that some of the physicochemical nanoparticle characteristics play in the uptake pathway preferentially used by the nanoparticles to gain access and deliver their cargo inside the cell. Keywords: nanoparticles; endocytosis; clathrin; caveolin; macropinocytosis 1. Introduction Nanomedicine has become one of the most rapidly growing areas of research in the biomedical field during the last years. Nanoparticles (NPs) and submicron particles (named both from this point as NPs to abbreviate) are defined as materials with nanometric sizes (1–100 and 100–1000 nm, respectively) that interact with biological systems in an unusual way because of their high surface to volume ratio. This property, combined with the possibility of modifying their peripheral chemical groups to achieve multitasking properties, provides NPs compounds with a very high potential for diagnostic and therapeutic applications in nanomedicine. -

From the Biology of Caveolin-3 to Human Diseases
European Journal of Human Genetics (2010) 18, 137–145 & 2010 Macmillan Publishers Limited All rights reserved 1018-4813/10 $32.00 www.nature.com/ejhg PRACTICAL GENETICS In association with Caveolinopathies: from the biology of caveolin-3 to human diseases In muscle tissue the protein caveolin-3 forms caveolae – flask-shaped invaginations localized on the cytoplasmic surface of the sarcolemmal membrane. Caveolae have a key role in the maintenance of plasma membrane integrity and in the processes of vesicular trafficking and signal transduction. Mutations in the caveolin-3 gene lead to skeletal muscle pathology through multiple pathogenetic mechanisms. Indeed, caveolin-3 deficiency is associated to sarcolemmal membrane alterations, disorganization of skeletal muscle T-tubule network and disruption of distinct cell-signaling pathways. To date, there have been 30 caveolin-3 mutations identified in the human population. Caveolin-3 defects lead to four distinct skeletal muscle disease phenotypes: limb girdle muscular dystrophy, rippling muscle disease, distal myopathy, and hyperCKemia. In addition, one caveolin-3 mutant has been described in a case of hypertrophic cardiomyopathy. Many patients show an overlap of these symptoms and the same mutation can be linked to different clinical phenotypes. This variability can be related to additional genetic or environmental factors.This review will address caveolin-3 biological functions in muscle cells and will describe the muscle and heart disease phenotypes associated with caveolin-3 mutations. In brief Hypertrophic Cardiomyopathy, 4 patients affected by Long QT Syndrome and 3 infants died from Sudden Infant Death In skeletal muscle tissue, the membrane protein Caveolin-3 Syndrome. regulates sarcolemmal stability and modulates the activity of Many patients show such an overlap of the four skeletal muscle different signalling pathways. -

A Novel SNAP25–Caveolin Complex Correlates with the Onset of Persistent Synaptic Potentiation
The Journal of Neuroscience, August 15, 2000, 20(16):5997–6006 A Novel SNAP25–Caveolin Complex Correlates with the Onset of Persistent Synaptic Potentiation Janice E. A. Braun1 and Daniel V. Madison2 1Department of Physiology and Biophysics, Neuroscience Research Group, The University of Calgary, Calgary, Alberta, Canada T2N 4N1, and 2Department of Molecular and Cellular Physiology, Stanford University Medical Center, Stanford, California 94305-5428 We have identified synaptic protein complexes in intact rat hip- correspond to complexes identified previously in vitro. After the pocampal slices using the rapid chemical cross-linking reagent induction of persistent synaptic potentiation, an abundant 40 paraformaldehyde. Cellular proteins were rapidly cross-linked, kDa SNAP25–caveolin1 complex was observed. The SNAP25– solubilized, separated electrophoretically by SDS-PAGE, and caveolin1 complex was not abundant in control slices and, there- then identified immunologically. Multiple complexes containing fore, represents the first demonstration of a reorganization of syntaxin, the synaptosomal-associated protein of 25 kDa protein complexes in intact hippocampal slices during the induc- (SNAP25), and vesicle-associated membrane protein (VAMP) tion of synaptic potentiation. The interaction between caveolin1 were observed to coexist in a single hippocampal slice including and SNAP25 was confirmed biochemically by demonstration of a 100 kDa cross-linked protein complex that exhibited the same the association of caveolin with recombinant-immobilized electrophoretic migration as a member of the previously identi- SNAP25 and by the coimmunoprecipitation of SNAP25 using fied SDS-resistant soluble N-ethylmaleimide-sensitive fusion at- caveolin-specific antisera. Caveolin1, like SNAP25, was ob- tachment protein receptor “core” of the 20 S complex. A VAMP– served to be abundant in isolated hippocampal nerve terminals synaptophysin complex, reported previously in vitro, was also (synaptosomes). -

Role of Caveolin-3 and Caveolae Mechanics in Muscle Pathophysiology Melissa Dewulf
Role of caveolin-3 and caveolae mechanics in muscle pathophysiology Melissa Dewulf To cite this version: Melissa Dewulf. Role of caveolin-3 and caveolae mechanics in muscle pathophysiology. Cellular Biol- ogy. Université Paris-Saclay, 2018. English. NNT : 2018SACLS067. tel-02145218 HAL Id: tel-02145218 https://tel.archives-ouvertes.fr/tel-02145218 Submitted on 2 Jun 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Thèse de doctorat de l'Université Paris-Saclay préparée à l’Université Paris-Saclay École doctorale n°568 Signalisations et réseaux intégratifs en biologie (Biosigne) Spécialité de doctorat: Biologie cellulaire et moléculaire Thèse présentée et soutenue à Paris, le 29 mars 2018, par Melissa Dewulf Role of caveolin-3 and caveolae mechanics in the muscle pathophysiology Composition du Jury : Pr. Karim Benihoud Président du jury Dr. Soazig Le lay Rapporteur Dr. Edgar Gomes Rapporteur Dr. Julie Dam Examinateur Dr. Stéphane Vassilopoulos Examinateur Dr. Christophe Lamaze Directeur de thèse ! ! ! ! ! ! ! « Et voilà, on a les droits ! » François Pignon Acknowledgements The last four years were probably the most challenging but also the most exciting of my life, and it is time for me to thank many people who were involved, in so many different ways, in this adventure.