Downloaded from the NCBI Database in in the Background Population Was Determined
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Comparison of Gene Expression Profiles Between Dental Pulp and Periodontal Ligament Tissues in Humans
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 40: 647-660, 2017 Comparison of gene expression profiles between dental pulp and periodontal ligament tissues in humans AI-XIU GONG1*, JING-HAN ZHANG2*, JING LI1, JUN WU3, LIN WANG4 and DENG-SHUN MIAO3 Departments of 1Stomatology, and 2Neonatology, Children's Hospital of Nanjing Medical University, Nanjing, Jiangsu 210008; 3State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University; 4Jiangsu Key Laboratory of Oral Diseases, Department of Orthodontics, School of Stomatology, Nanjing Medical University, Nanjing, Jiangsu 210029, P.R. China Received March 16, 2016; Accepted June 16, 2017 DOI: 10.3892/ijmm.2017.3065 Abstract. There are anatomical and functional differ- DP samples. The gene expression profiles provide novel insight ences between human dental pulp (DP) and periodontal into the characterization of DP and PDL tissues, and contribute ligament (PDL). However, the molecular biological differ- to our understanding of the potential molecular mechanisms of ences and function of these tissues are poorly understood. dental tissue mineralization and regeneration. In the present study, we employed a cDNA microarray array to screen for differentially expressed genes (DEGs) between Introduction human DP and PDL tissues, and used the online software WebGestalt to perform the functional analysis of the DEGs. Dental pulp (DP) tissue, termed as the ‘ectomesenchyme’, is In addition, the STRING database and KEGG pathway derived from ectodermal cells that grow on the periphery of analysis were applied for interaction network and pathway the neural tube, migrate to the oral position and then differen- analysis of the DEGs. -

Integrin Alpha-5 Silencing Leads to Myofibroblastic Differentiation In
TAJ0010.1177/2040622320936023Therapeutic Advances in Chronic DiseaseGE Shochet 936023research-article20202020 Therapeutic Advances in Chronic Disease Original Article Ther Adv Chronic Dis Integrin alpha-5 silencing leads to 2020, Vol. 11: 1–12 DOI:https://doi.org/10.1177/2040622320936023 10.1177/ myofibroblastic differentiation in IPF- 2040622320936023https://doi.org/10.1177/2040622320936023 © The Author(s), 2020. Article reuse guidelines: derived human lung fibroblasts sagepub.com/journals- permissions Gali Epstein Shochet* , Elizabetha Brook*, Becky Bardenstein-Wald, Hanna Grobe, Evgeny Edelstein, Lilach Israeli-Shani and David Shitrit Abstract Background and objective: The term ‘fibroblast’ covers a heterogeneous cell population in idiopathic pulmonary fibrosis (IPF). The fibroblasts are considered as main effector cells, because they promote disease progression by releasing exaggerated amounts of extracellular matrix proteins and modifying cell microenvironment. As IPF-derived human lung fibroblasts (IPF-HLFs) were shown to express higher levels of integrin alpha-5 (ITGA5) than normal derived HLFs (N-HLFs), we explored the importance of ITGA5 to IPF progression. Methods: IPF-HLF and N-HLF primary cultures were established. ITGA5 was silenced by specific small interfering RNA (siRNA)s and its effects on cell phenotype (e.g. cell number, size, cell death, migration) and gene expression (e.g. RNA sequencing, quantitative polymerase chain reaction [qPCR], western blot and immunofluorescence) were tested. Specific integrin expression was evaluated in IPF patient formalin-fixed paraffin embedded sections by immunohistochemistry (IHC). Results: ITGA5-silencing resulted in reduced IPF-HLF proliferation rates and cell migration (p < 0.05), as well as elevated cell death. transforming growth factor beta (TGF-β) targets (e.g. Fibronectin (FN1), Matrix metalloproteinase 2 (MMP2), TGFB1) were surprisingly elevated following ITGA5 silencing (p < 0.05). -

Integrin and Gene Network Analysis Reveals That ITGA5 and ITGB1 Are Prognostic in Non-Small-Cell Lung Cancer
Journal name: OncoTargets and Therapy Article Designation: Original Research Year: 2016 Volume: 9 OncoTargets and Therapy Dovepress Running head verso: Zheng et al Running head recto: ITGA5 and ITGB1 are prognostic in NSCLC open access to scientific and medical research DOI: http://dx.doi.org/10.2147/OTT.S91796 Open Access Full Text Article ORIGINAL RESEARCH Integrin and gene network analysis reveals that ITGA5 and ITGB1 are prognostic in non-small-cell lung cancer Weiqi Zheng Background: Integrin expression has been identified as a prognostic factor in non-small-cell Caihui Jiang lung cancer (NSCLC). This study was aimed at determining the predictive ability of integrins Ruifeng Li and associated genes identified within the molecular network. Patients and methods: A total of 959 patients with NSCLC from The Cancer Genome Atlas Department of Radiation Oncology, Guangqian Hospital, Quanzhou, Fujian, cohorts were enrolled in this study. The expression profile of integrins and related genes were People’s Republic of China obtained from The Cancer Genome Atlas RNAseq database. Clinicopathological characteristics, including age, sex, smoking history, stage, histological subtype, neoadjuvant therapy, radiation therapy, and overall survival (OS), were collected. Cox proportional hazards regression models as well as Kaplan–Meier curves were used to assess the relative factors. Results: In the univariate Cox regression model, ITGA1, ITGA5, ITGA6, ITGB1, ITGB4, and ITGA11 were predictive of NSCLC prognosis. After adjusting for clinical factors, ITGA5 (odds ratio =1.17, 95% confidence interval: 1.05–1.31) andITGB1 (odds ratio =1.31, 95% confidence interval: 1.10–1.55) remained statistically significant. In the gene cluster network analysis, PLAUR, ILK, SPP1, PXN, and CD9, all associated with ITGA5 and ITGB1, were identified as independent predictive factors of OS in NSCLC. -
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/7223/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-3767223.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Pig Extracellular Matrix & Adhesion Molecules Cat. no. 330231 PASS-013ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Pig Extracellular Matrix & Adhesion Molecules RT² Profiler PCR Array profiles the expression of 84 genes important for cell-cell and cell-matrix interactions. Cells attach to proteoglycans and glycoproteins (such as fibronectin, laminin, and collagen) in the extracellular matrix (ECM) substratum via adhesion molecules on their cell surface to define tissue shape, structure, and function. Making and breaking cellular contacts with other cells and the ECM play critical roles in normal processes such as cell growth, division, differentiation, and migration. -

Integrin A8 Is Abundant in Human, Rat, and Mouse Trophoblasts
Original Article Reproductive Sciences 2017, Vol. 24(10) 1426-1437 ª The Author(s) 2017 Integrin a8 Is Abundant in Human, Rat, and Reprints and permission: sagepub.com/journalsPermissions.nav Mouse Trophoblasts DOI: 10.1177/1933719116689597 journals.sagepub.com/home/rsx Sebastian Herdl1, Hanna Huebner2, Gudrun Volkert, PhD1, Ines Marek, MD1, Carlos Menendez-Castro, MD1, Stephanie C. Noegel, MD1, Matthias Ruebner, PhD2, Wolfgang Rascher, MD1, Andrea Hartner, PhD1, and Fabian B. Fahlbusch, MD1 Abstract Objective: Integrins exert regulatory functions in placentogenesis. Null mutation of certain integrin a subunits leads to placental defects with subsequent fetal growth restriction or embryonic lethality in mice. So far, the placental role of a8 integrin remains to be determined. Methods: Localization of a8 integrin and its ligands, fibronectin (FN) and osteopontin (OPN), was studied by immunohistochemistry in human, rat, and mouse placenta. The vascularization of the placental labyrinth layer of a8 integrin- deficient mice was determined by CD31 staining. In humans, a8 integrin expression was assessed via real-time polymerase chain reaction in healthy placentas, in the placental pathologies such as intrauterine growth restriction (IUGR), preeclampsia, and HELLP-syndrome (hemolysis, elevated liver enzymes, low platelet count), as well as in primary extravillous trophoblasts (EVT) and villous trophoblasts. Results: In humans, a8 integrin was detected in first and third trimester syncytiotrophoblast and EVT. Although OPN showed the same localization, FN was observed in EVT only. No expressional changes in a8 integrin were detected in the placental pathologies studied. Rodent placenta showed a8 integrin expression in giant cells and in the labyrinth layer. The localization of OPN and FN, however, showed species-specific differences. -

Bioinformatics and Statistical Genomics
Genomic Med. (2008) 2:217–240 DOI 10.1007/s11568-009-9081-x ABSTRACTS Bioinformatics and statistical genomics Ó Human Genome Organisation (HUGO) International Limited 2009 001: A physical map of human Alu repeats cleavage responses to various drugs. Variations are mostly due to non-synon- by restriction endonucleases ymous single nucleotide polymorphisms (nsSNPs) some of which have been associated to certain diseases. However, large number of nsSNPs have not been associated to any of the known diseases and Murat A. Abdurashitov, Victor N. Tomilov, Valery A. Chernukhin, have remained uncharacterized. In addition to these, on-going genetic Sergey Kh Degtyarev studies and human population based studies have also been leading to SibEnzyme Ltd., 2/12 Ak. Timakov Str., Novosibirsk 630117, Russia the discovery of new SNPs. It is important to predict the effect of nsSNPs at the molecular level as well as at the physiological level. The Alu family of DNA repeats, which belongs to SINE group, is one In this study we have focused on the nsSNPs which occur on the of the most abundant and well characterized repetitive elements in transmembrane part of the human membrane transport proteins and try human genome. The total number of annotated Alu sequences is more to develop a tool which can classify uncharacterized nsSNPs into than 1 million copies and their fraction in genome is about 10%. benign and disease nsSNPs. In order to develop such a tool we have Earlier we have shown that visible bands on electrophoretical gels investigated and discovered a number of sequence-based properties after total human DNA digestion are formed mainly due to cleavage and features such as burial status, evolutionary characteristics etc., at of Alu repeats. -

Cell Adhesion Molecules Are Mediated by Photobiomodulation at 660 Nm in Diabetic Wounded Fibroblast Cells
cells Article Cell Adhesion Molecules Are Mediated by Photobiomodulation at 660 nm in Diabetic Wounded Fibroblast Cells Nicolette N. Houreld * ID , Sandra M. Ayuk and Heidi Abrahamse ID Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein, Johannesburg 2028, South Africa; [email protected] (S.M.A.); [email protected] (H.A.) * Correspondence: [email protected]; Tel.: +27-11-559-6833 Received: 9 March 2018; Accepted: 12 April 2018; Published: 16 April 2018 Abstract: Diabetes affects extracellular matrix (ECM) metabolism, contributing to delayed wound healing and lower limb amputation. Application of light (photobiomodulation, PBM) has been shown to improve wound healing. This study aimed to evaluate the influence of PBM on cell adhesion molecules (CAMs) in diabetic wound healing. Isolated human skin fibroblasts were grouped into a diabetic wounded model. A diode laser at 660 nm with a fluence of 5 J/cm2 was used for irradiation and cells were analysed 48 h post-irradiation. Controls consisted of sham-irradiated (0 J/cm2) cells. Real-time reverse transcription (RT) quantitative polymerase chain reaction (qPCR) was used to determine the expression of CAM-related genes. Ten genes were up-regulated in diabetic wounded cells, while 25 genes were down-regulated. Genes were related to transmembrane molecules, cell–cell adhesion, and cell–matrix adhesion, and also included genes related to other CAM molecules. PBM at 660 nm modulated gene expression of various CAMs contributing to the increased healing seen in clinical practice. There is a need for new therapies to improve diabetic wound healing. -

Integrin A4h1 Function Is Required for Cell Survival in Developing Retina
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Developmental Biology 276 (2004) 416–430 www.elsevier.com/locate/ydbio Integrin a4h1 function is required for cell survival in developing retina Sergiu T. Leua, Susan A.L. Jacquesa, Kevin L. Wingerda, Sherry T. Hikitaa, Erin C. Tolhursta, Jan L. Pringa, Derek Wiswella, Lisa Kinneya, Nichol L. Goodmana,1, David Y. Jacksonb, Dennis O. Clegga,* aNeuroscience Research Institute and Department of Molecular, Cellular and Developmental Biology, University of California, Santa Barbara, CA 93106, United States bDepartment of Bioorganic Chemistry, Genentech Inc., South San Francisco, CA 94080, United States Received for publication 10 April 2004, revised 31 August 2004, accepted 1 September 2004 Available online 1 October 2004 Abstract In the retina, integrins in the h1 family have been shown to be important in many phases of neuronal development, particularly neuroblast migration and axon outgrowth. However, the functions of specific integrin heterodimers are not well defined. In this study, we investigated the functions of h1 integrins in developing chicken retina by expression of a dominant-negative h1A construct using a replication-competent retrovirus. Inhibition of integrins using this approach resulted in alteration of cell morphology and increased apoptosis, but did not preclude migration and axon elongation. In an attempt to identify which specific h1 heterodimer was important, expression and function of the a4h1 heterodimer were also investigated. At early developmental stages, a4 protein and mRNA were detected in undifferentiated neuroblasts throughout the retina. At later stages, expression was confined to retinal ganglion cells (RGCs) and amacrine cells. -

A Promoter Polymorphism of the Alpha 8 Integrin Gene and the Progression of Autosomal-Dominant Polycystic Kidney Disease
Original Paper Nephron Clin Pract 2008;108:c169–c175 Received: March 23, 2007 DOI: 10.1159/000116887 Accepted: October 15, 2007 Published online: February 14, 2008 A Promoter Polymorphism of the Alpha 8 Integrin Gene and the Progression of Autosomal-Dominant Polycystic Kidney Disease a a a a Raoul Zeltner Karl F. Hilgers Roland E. Schmieder Markus Porst a b Bernd D. Schulze Andrea Hartner a b Department of Nephrology and Hypertension, and Children and Youth Hospital, University of Erlangen-Nuremberg, Erlangen , Germany Key Words failure was significantly different (p = 0.026) whereas age at Alpha 8 integrin ؒ Promoter ؒ Autosomal-dominant onset of hypertension, body surface area, 24-hour systolic polycystic kidney disease ؒ Association study ؒ End-stage and diastolic blood pressure did not differ. In kidneys of renal disease ADPKD, expression of alpha 8 integrin is increased and found de novo in cystic epithelia. Conclusion: A polymorphism of the ITGA8 promoter modifies the progression of renal failure Abstract in ADPKD. Copyright © 2008 S. Karger AG, Basel Background/Aims: Dysregulation of integrins is a feature of tissue remodeling in autosomal-dominant polycystic kidney disease (ADPKD). The alpha 8 beta 1 integrin (␣ 8  1) affects kidney development and the susceptibility to renal injury in Introduction mice. We investigated whether the –414 T/C polymorphism in the promoter region of the alpha 8 integrin chain gene The alpha 8 beta 1 integrin ( ␣ 8  1) belongs to a family (ITGA8) is associated with the progression of renal disease in of heterodimeric cell-matrix receptors consisting of one ADPKD. Methods: Genotyping for the –414 T/C polymor- ␣ and one  subunit [1, 2] . -
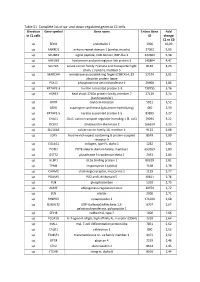
Table S1. Complete List of Up- and Down-Regulated Genes in C1 Cells
Table S1. Complete list of up- and down-regulated genes in C1 cells. Direction Gene symbol Gene name Entrez Gene Fold in C1 cells ID change C1 vs C0 up EDN1 endothelin 1 1906 10,09 up ANKRD1 ankyrin repeat domain 1 (cardiac muscle) 27063 5,95 up SCUBE3 signal peptide, CUB domain, EGF-like 3 222663 5,38 up HAPLN3 hyaluronan and proteoglycan link protein 3 145864 4,47 up SLC7A5 solute carrier family 7 (amino acid transporter light 8140 4,23 chain, L system), member 5 up MARCH4 membrane-associated ring finger (C3HC4) 4, E3 57574 3,91 ubiquitin protein ligase up PSAT1 phosphoserine aminotransferase 1 29968 3,86 up KRTAP2-3 keratin associated protein 2-3 730755 3,76 up HSPB7 heat shock 27kDa protein family, member 7 27129 3,74 (cardiovascular) up OXTR oxytocin receptor 5021 3,52 up ASNS asparagine synthetase (glutamine-hydrolyzing) 440 3,39 up KRTAP1-5 keratin associated protein 1-5 83895 3,27 up CHAC1 ChaC, cation transport regulator homolog 1 (E, coli) 79094 3,22 up DCLK2 doublecortin-like kinase 2 166614 3,15 up SLC16A4 solute carrier family 16, member 4 9122 3,08 up LGR5 leucine-rich repeat containing G protein-coupled 8549 2,99 receptor 5 up COL4A1 collagen, type IV, alpha 1 1282 2,94 up POTEI POTE ankyrin domain family, member I 653269 2,89 up GSTT2 glutathione S-transferase theta 2 2953 2,84 up ULBP1 UL16 binding protein 1 80329 2,81 up TPM1 tropomyosin 1 (alpha) 7168 2,78 up CHRM2 cholinergic receptor, muscarinic 2 1129 2,77 up PDLIM5 PDZ and LIM domain 5 10611 2,76 up PLN phospholamban 5350 2,75 up ADIRF adipogenesis regulatory factor -

1 Imipramine Treatment and Resiliency Exhibit Similar
Imipramine Treatment and Resiliency Exhibit Similar Chromatin Regulation in the Mouse Nucleus Accumbens in Depression Models Wilkinson et al. Supplemental Material 1. Supplemental Methods 2. Supplemental References for Tables 3. Supplemental Tables S1 – S24 SUPPLEMENTAL TABLE S1: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S2: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S3: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S4: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Isolation Model (p<0.001) SUPPLEMENTAL TABLE S5: Genes Demonstrating Common Altered Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S6: Genes Demonstrating Increased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S7: Genes Demonstrating Decreased Repressive DimethylK9/K27-H3 Methylation in the Social Defeat and Social Isolation Models (p<0.001) SUPPLEMENTAL TABLE S8: Genes Demonstrating Increased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S9: Genes Demonstrating Decreased Phospho-CREB Binding in the Social Defeat Model (p<0.001) SUPPLEMENTAL TABLE S10: Genes Demonstrating Increased Phospho-CREB Binding in the Social