Koppel Georgetown 0076D 12
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hydrogenation of Styrene Oxide to 2-Phenylethanol Over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method
Hindawi Publishing Corporation International Journal of Chemical Engineering Volume 2014, Article ID 406939, 6 pages http://dx.doi.org/10.1155/2014/406939 Research Article Hydrogenation of Styrene Oxide to 2-Phenylethanol over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method Sunil K. Kanojiya,1 G. Shukla,1 S. Sharma,1 R. Dwivedi,2 P. Sharma,2 R. Prasad,2 M. Satalkar,3 and S. N. Kane3 1 IPCA Laboratories Ltd., Indore 452003, India 2 School of Chemical Sciences, DA University, Indore 452001, India 3 Magnetic Materials Laboratory, School of Physics, DA University, Indore 452001, India Correspondence should be addressed to R. Prasad; [email protected] Received 14 February 2014; Accepted 20 May 2014; Published 17 July 2014 Academic Editor: Moshe Sheintuch Copyright © 2014 Sunil K. Kanojiya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Nanocrystalline nickel prepared by glycol reduction method and characterized by XRD and magnetic measurements has been used as a catalyst for hydrogenation of styrene oxide to 2-phenylethanol. Effect of process variables such as particle size of the catalyst, temperature, and pressure have been optimized to achieve a maximum conversion of 98% of styrene oxide with 99% selectivity towards 2-phenylethanol. The structure of the transition state has been computed employing density functional theory −1 and using Gaussian 09 suite. The enthalpy of reaction (Δ) and activation energy ()arecalculatedtobe85.3kcal⋅mol and −1 123.03 kcal⋅mol , respectively. A tentative mechanism for the reaction is proposed according to which atomized hydrogen and styrene oxide react together over the catalyst surface to produce 2-phenylethanol. -

Chem 353 Derivative Tables
ALCOHOLS Derivatives Mp/ oC Compound Bp /oC 3,5-Dinitro- Phenylurethane Naphthyl- (Mp / oC) benzoate urethane 4-Methylbenzyl alcohol --- (60) 118 79 --- Diphenylmethanol --- (69) 141 136 --- Ethyl p-hydroxybenzoate --- (116) * * * Methyl p-hydroxybenzoate --- (130) * * * Ethanol 78 93 52 79 2-Propanol 83 122 88 106 1-Propanol 97 74 51 80 2-Butanol 99 75 65 97 2-Methyl-1-propanol 108 86 86 104 3-Methyl-2-butanol 113 76 68 110 1-Butanol 116 64 63 71 3-Pentanol 116 101 48 95 2-Pentanol 119 61 --- (oil) 76 2-Chloroethanol 129 92 51 101 2-Methyl-1-butanol 129 70 31 82 3-Methyl-1-butanol 132 61 55 68 4-Methyl-2-pentanol 132 65 143 88 3-Methyl-2-pentanol 134 43 --- 72 1-Pentanol 138 46 46 68 2-Methyl-1-pentanol 148 51 --- 75 2-Ethyl-1-butanol 149 52 --- --- 1-Hexanol 156 58 42 59 Cyclohexanol 161 112 82 129 2-Octanol 179 32 114 63 1-Phenylethanol 203 95 94 106 Benzyl alcohol 205 112 78 134 2-Phenylethanol 219 108 79 119 Ethyl o-hydroxybenzoate 234 * * * Cinnamyl alcohol 257 (33) 121 90 114 4-Methoxybenzyl alcohol 260 (25) --- 92 --- * These compounds are also esters, see that table for derivatives. --- No data available. ESTERS Derivative Mp/ oC Compound Bp /oC Carboxylic (Mp / oC) Acid Methyl p-methylbenzoate 223 (30) 177 Methyl cinnamate 261 (35) 133 Methyl 4-methoxybenzoate 245 (49) 184 Ethyl p-nitrobenzoate --- (57) 241 Methyl p-nitrobenzoate --- (95) 241 Ethyl p-hydroxybenzoate --- (116) 213 Methyl p-hydroxybenzoate --- (130) 213 Methyl benzoate 198 121 Methyl o-toluate 213 102 Ethyl benzoate 213 121 Diethyl succinate 216 190 Methyl phenylacetate 218 76 Methyl salicylate 224 157 Methyl o-chlorobenzoate 230 140 Ethyl salicylate 234 157 Ethyl p-toluate 241 177 Isopropyl salicylate 255 157 Ethyl o-chlorobenzoate 255 140 Dimethyl suberate 268 141 Ethyl 4-methoxybenzoate 270 184 Ethyl cinnamate 271 133 Diethyl phthalate 296 230 --- No data available. -

Ch3ch2cch(Ch3)2 O Ch2ch2cho Ch3cch2ch2ch2cch2ch3
Chem 226 — Problem Set #9 — “Fundamentals of Organic Chemistry,” 4th edition, John McMurry. Chapter 9 2. Name the following aldehydes and ketones. O (a) (b) CH2CH2CHO CH3CH2CCH(CH3)2 H CH3 (c) O O (d) H C O CH3CCH2CH2CH2CCH2CH3 H (a) 2-methyl-3-pentanone, (b) 3-phenylpropanal, (c) 2,6-octanedione, (d) (1R,2R)-2-methylcyclohexanecarbaldehyde 4. How could you prepare pentanal from the following starting materials? (a) 1-pentanol, (b) CH3CH2CH2CH2COOH, (c) 5-decene PCC (a) CH3CH2CH2CH2CH2OH CH3CH2CH2CH2CHO CH2Cl2 O 1. LiAlH4 CH3CH2CH2CH2C OH CH3CH2CH2CH2CH2OH H O+ (b) 2. 3 PCC CH3CH2CH2CH2CH2OH CH3CH2CH2CH2CHO CH2Cl2 KMnO4 CH3CH2CH2CH2CH CHCH2CH2CH2CH3 + H3O O LiAlH (c) 1. 4 CH3CH2CH2CH2C OH + CH3CH2CH2CH2CH2OH 2. H3O PCC CH3CH2CH2CH2CH2OH CH3CH2CH2CH2CHO CH2Cl2 5. How could you prepare 2-hexanone from the following starting materials? (a) 2-hexanol, (b) 1-hexyne, (c) 2-methyl-1-hexene OH O K2CrO7 (a) CH3CH2CH2CH2CHCH3 CH3CH2CH2CH2CCH3 O H2SO4 (b) CH3CH2CH2CH2C CH CH3CH2CH2CH2CCH3 HgSO4 H2O CH3 O KMnO4 (c) CH3CH2CH2CH2C CH2 CH3CH2CH2CH2CCH3 + H3O (b) The enol that initially forms here undergoes keto-enol tautomerism. Terminal alkynes give methyl ketones; they follow Markovnikov’s rule. (c) Potassium permanganate under basic conditions would hydroxylate the alkene to form a vicinal diol, but under acidic conditions it completely cleaves the double bond. The =CH2 group would become carbon dioxide. 6. How would you carry out the following transformations? More than one step may be required. (a) 3-hexene 3-hexanone (b) benzene 1-phenylethanol H OH H2O (a) CH3CH2CH CHCH2CH3 CH3CH2CH CHCH2CH3 H SO 2 4 H O K2Cr2O7 CH3CH2CH CCH2CH3 O CH3CCl (b) C CH3 AlCl3 O H NaBH4 C CH3 OH 9. -

Green Chemistry : Greener Alternatives to Synthetic Organic Transformations
Green Chemistry Greener Alternatives to Synthetic Organic Transformations V.K. Ahluwalia Alpha Science International Ltd. Oxford, U.K. Contents Preface v Parti 1. Introduction 1.1-1.10 1.1 Principles of Green Chemistry 1.1 1.2 How to Plan a Green Synthesis 1.2 Part II Green Alternatives to Synthesis Organic Transformations 2. Aqueous Phase Transformations 2.1-2.48 2.1 p-Acetylaminophenol (Tylenol) 2.1 2.2 3-Aminopyridine 2.2 2.2a Anthranilic Acid 2.3 2.3 Benzilic Acid 2.3 2.4 Benzoin 2.5 2.5 Benzotriazole 2.6 2.6 2-Benzoyl-3,5-dimethylbenzofuran 2.7 2.7 n-Butyl Bromide 2.8 2.8 Tert.Butylchloride 2.8 2.9 Chalcone (Benzalacetophenone) 2.9 2.10 Cycloheptanone 2.10 2.11 2,3-Dihydroxy Anisole (Pyrigallol Monomethyl-ether) 2.12 2.12 2,4-Dihydroxybenzoic acid (P-resorcylic Acid) 2.13 2.13 3,4-Dimethoxyphenol 2.14 2.14 2,3-Dimethyl-l-phenylpyrazol-5-one 2.15 2.15 3, 5-Dimethylpyrazole 2.16 2.16 5,5-Diphenylhydantoin 2.17 2.17 Endo-cis-1,4-endoxo-A5-cyclohexene-2,3-dicarboxylic Acid 2.18 2.18 p-Ethoxyacetanilide (Phenacetin) 2.19 2.19 6-Ethoxycarbonyl-3,5-diphenyI-2-cyclohexenone 2.20 2.20 HeteroDiels-AlderAdduct 2.22 2.21 Hippuric Acid (Benzoyl Glycine) 2.22 Viii Contents 2.22 Hydantion 2.23 2.25 3-Hydroxy-3-phenyl-2-methylene Proponamide 2.24 2.24 Iodoform 2.25 2.25 Inodole 2.26 2.26 3-(p-Methoxyphenyl)-2H-1,4-Benzoxazine 2.27 2.27 3-Methylcyclopent-2-enone 2.27 2.28 2-(2'-Methlindol-3yl)-1,4-benzoquinone 2.28 2.29 2-methyl-2-(3-oxobutyl)-1,3-cyclopentanedione 2.29 2.30 P-Naphthyl Acetate 2.30 2.31 (5-Naphthyl Methyl Ether (Nerolin) 2.31 2.32 -

Inhibition of Growth, Synthesis, and Permeability in Neurospora Crassa by Phenethyl Alcohol
JOURNAL OF BACTERIOLOGY, JUly, 1965 Vol. 90, No. I Copyright © 1965 American Society for Microbiology Printed in U.S.A. Inhibition of Growth, Synthesis, and Permeability in Neurospora crassa by Phenethyl Alcohol GABRIEL LESTER Department of Biology, Reed College, Portland, Oregon Received for publication 22 January 1965 ABSTRACT LESTER, GABRIEL (Reed College, Portland, Ore.). Inhibition of growth, synthesis, and permeability in Neurospora crassa by phenethyl alcohol. J. Bacteriol. 90: 29-37. 1965.- Inhibition of the growth of Neurospora crassa in still culture was detected at 0.05% and was complete at a level of 0.2% phenethyl alcohol (PEA). Benzyl alcohol was less in- hibitory, and 3-phenyl-1-propanol and phenol were more inhibitory, than PEA; benzyl- amine and phenethylamine were less inhibitory than the analogous hydroxylated com- pounds. Inhibition by PEA was not reversed by synthetic mixtures of purines and pyrimidines or vitamins, or by casein digests, yeast extract, or nutrient broth. The germination of conidia was inhibited by PEA, but after an exposure of 8.5 hr no loss of viability was observed. The addition of PEA to growing shake cultures caused a simul- taneous inhibition of growth and of the syntheses of ribonucleic and deoxyribonucleic acids and protein; the relationships of these compounds to mycelial dry weight and to one another were constant in growing mycelia, and PEA did not significantly affect these relationships. PEA partially inhibited the uptake of glucose, but severely re- stricted the accumulation of L-leucine, L-tryptophan, or a-aminoisobutyric acid in germinated conidia. The efflux of a-aminoisobutyric acid from germinated conidia was somewhat enhanced by PEA, but this effect was not so pronounced as the (complete) inhibition of a-aminoisobutyric acid accumulation by PEA. -

United States Patent (19) 11 4,267,375 Maasbol Et Al
United States Patent (19) 11 4,267,375 Maasbol et al. 45 May 12, 1981 54 PREPARATION OF THIOETHERS OTHER PUBLICATIONS 75) Inventors: Alfred G. Maasbol, Hamburg, Fed. I. Ruderman et al., J. Amer. Chem. Soc., 71, pp. Rep. of Germany; Lothar G. Dulog, 2264-2265, (1949). St. Martens Latem, Belgium Morrisson and Boyd, Organic Chemistry, 2nd edition, (1967), pp. 29–30. 73) Assignee: s.a. Texaco Belgium in.v., Brussels, T. Todsen et al., J. Amer. Chem. Soc., 72, pp. Belgium 4000-4002, (1950). Berichte Deutsch. Chemie, vol. 1, pp. 587-591, (1935), (21) Appl. No.: 945,273 Berlin. 22 Filed: Sep. 25, 1978 D. Gregg et al., J. Org. Chem., pp. 246-252, (1950). M. Malinovskii, Epoxides and Their Derivatives, pp. Related U.S. Application Data 131-136, (1965), Jerusalem, Daniel Davey & Co. Primary Examiner-Glennon H. Hollrah 63 Continuation of Ser. No. 703,045, Jul. 6, 1976, aban Assistant Examiner-M. C. Eakin doned. Attorney, Agent, or Firm-Carl G. Ries; Robert A. 30 Foreign Application Priority Data Kulason; Carl G. Seutter Nov. 19, 1975 GB United Kingdom ............... 47582/75 57 ABSTRACT .. 51 Int. Cl. ............................................ CO7C 149/30 Thioethers may be prepared by reacting a thiol, such as thiophenol, with an alcohol (having electron donor 52 U.S. C. ......................................... 568/57; 568/58 groups in the alpha or beta position to its hydroxyl 58 Field of Search ........................ 260/609 E, 609 R group) such as phenyl-1-hydroxy-phenethylsulfide. Re 56) References Cited action is carried out in the presence of a Lewis Acid U.S. PATENT DOCUMENTS metal halide, typically zinc chloride. -
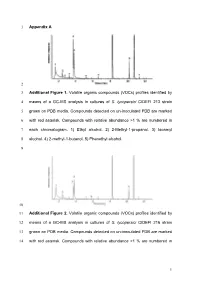
Appendix a 1 2 Additional Figure 1. Volatile Organic Compounds (Vocs
1 Appendix A 2 3 Additional Figure 1. Volatile organic compounds (VOCs) profiles identified by 4 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 213 strain 5 grown on PDB media. Compounds detected on un-inoculated PDB are marked 6 with red asterisk. Compounds with relative abundance >1 % are numbered in 7 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 8 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 9 10 11 Additional Figure 2. Volatile organic compounds (VOCs) profiles identified by 12 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 216 strain 13 grown on PDB media. Compounds detected on un-inoculated PDB are marked 14 with red asterisk. Compounds with relative abundance >1 % are numbered in 1 15 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 16 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 6) Furfuryl alcohol. 17 18 19 Additional Figure 3. Volatile organic compounds (VOCs) profiles identified by 20 means of a GC-MS analysis in cultures of F. fulva CIDEFI 300 strain grown on 21 PDB media. Compounds detected on un-inoculated PDB are marked with red 22 asterisk. Compounds with relative abundance >1 % are numbered in each 23 chromatogram. 6) Furfuryl alcohol. 7) Acetone. 8) Methyl trimethylacetate. 9) 24 Isoamyl alcohol. 10) 1-Octene. 11) 3-Hexanone, 4-methyl-. 12) Styrene. 13) 3- 25 Octanone. 14) Hexanoic acid, 2 ethyl-, methyl ester. 15) 2-Nonanone. 16) 26 Phenethyl alcohol. 17) No identified Nist05. 27 2 28 29 Additional Figure 4. -

Chemicals That Form Explosive Levels of Peroxides Without Concentration (Safe Storage Time After Opening - 3 Months) Chemical CAS # Synonym State Ref
Group A- Chemicals that form explosive levels of peroxides without concentration (Safe storage time after opening - 3 months) Chemical CAS # Synonym State Ref. 000106- Butadiene(1,3) 1,3-Butadiene gas 4 99-0 000126- 2-Chloro-1,3- Chloroprene (1,3) liquid 4 99-8 butadiene 000821- Divinyl acetylene 1,5-Hexadien- 3-yne liquid 5 08-9 000108- Isopropyl ether liquid 5 20-3 000116- Tetrafluoroethylene gas 4 14-3 000109- Vinyl ether Divinyl ether liquid 5 93-3 000075- 1,1- Vinylidene chloride liquid 5 35-4 Dichloroethylene Group B-Chemicals that form explosive levels of peroxides on concentration (Safe storage time after opening - 12 months) Chemical CAS # Synonym State Ref. 000105- Acetal liquid 5 57-7 000075- Acetaldehyde liquid 4 07-0 000100- Benzyl alcohol liquid 4 51-6 000078- 2-Butanol liquid 4 92-2 000108- Cyclohexanol liquid 4 93-0 000110- Cyclohexene liquid 5 83-8 000822- 2-Cyclohexen-1-ol liquid 4 67-3 000142- Cyclopentene liquid 5 29-0 000091- Decahydronaphthalene liquid 4 17-8 000460- Diacetylene gas 5 12-8 000077- Dicyclopentadiene liquid 5 73-6 Diethylene glycol 000111- Diglyme liquid 5 dimethyl ether 96-6 000123- Dioxane 1,4-Dioxane liquid 5 91-1 Ethylene glycol 000110- Glyme liquid 5 dimethyl ether 71-4 000060- Ethyl ether Diethyl ether liquid 5 29-7 000110- Furan liquid 5 00-9 000589- 4-Heptanol liquid 4 55-9 000626- 2-Hexanol liquid 4 93-7 000098- Isopropyl benzene Cumene liquid 5 82-8 000074- Methyl acetylene Propyne gas 5 99-7 000123- 3-Methyl-1-butanol Isoamyl alcohol liquid 4 51-3 000096- Methyl cyclopentane liquid 5 37-7 -

CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic Anhydride Liquid
CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic anhydride liquid Acetaminophen Synthesis p-aminophenol solid Alcohols to Alkyl chlorides 2-pentanol liquid Alcohols to Alkyl chlorides Hydrochloric acid 12 M Alcohols to Alkyl chlorides Sodium carbonate solid Alcohols to Alkyl chlorides Hydrobromic acid 48% w/v Alcohols to Alkyl chlorides Sodium sulfate anhydrous solid Alcohols to Alkyl chlorides sec-phenethyl alcohol liquid Alcohols to Alkyl chlorides Benzyl alcohol liquid Alcohols to Alkyl chlorides t-butanol liquid Alcohols to Alkyl chlorides 1-pentanol liquid Alcohols to Alkyl chlorides Sodium carbonate 10% w/v Diels Alder Reaction 2,3-dimethyl-1,3-butadiene liquid Diels Alder Reaction Maleic anhydride solid Diels Alder Reaction Ethanol 95% Liquid Diels Alder Reaction Hexane liquid Diels Alder Reaction Cyclohexane liquid Diels Alder Reaction Calcium chloride solid Esterification methanol liquid Esterification Sodium carbonate 10% w/v Esterification 1-propanol liquid Esterification 1-butanol liquid Esterification trans-cinnamic acid solid Esterification Isoamyl alcohol liquid Esterification Isopropyl alcohol liquid Esterification Benzyl alcohol liquid Esterification Sulfuric acid conc. 18 M Esterification 1-pentanol liquid Esterification Isobutyl alcohol liquid Esterification Ethanol 95% liquid Page 1 of 3 Experiment Title Chemical Name Concentration Extraction of Beta Carotene Cyclohexane liquid Extraction of Beta Carotene Beta carotene UV -

Phenethyl Alcohol Is an Effective Non-Traditional Preservative Agent for Cosmetic Preparations
Online - 2455-3891 Vol 10, Issue 8, 2017 Print - 0974-2441 Research Article PHENETHYL ALCOHOL IS AN EFFECTIVE NON-TRADITIONAL PRESERVATIVE AGENT FOR COSMETIC PREPARATIONS SASITHORN SIRILUN1, CHAIYAVAT CHAIYASUT1, BHAGAVATHI SUNDARAM SIVAMARUTHI1, SARTJIN PEERAJAN2, NAPHATSORN KUMAR3, PERIYANAINA KESIKA1* 1Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand. 2Health Innovation Institute, Chiang Mai 50200, Thailand. 3School of Cosmetic Science, Mae Fah Luang University, Chiang Rai 57100, Thailand. Email: [email protected]; [email protected] Received: 19 March 2017, Revised and Accepted: 26 April 2017 ABSTRACT Objective: Preservatives are used in the cosmetic products to protect the potential growth of microbes, therefore, to prolong the shelf life of products, and to protect the consumer from infections. However, several preservatives can cause various health problems, and the safety profiles of those preservatives are still unclear. Many natural substances are used in the cosmetic products to substitute the traditional preservatives. The present study deals with the evaluation of conservative nature of phenethyl alcohol (PEA) in three cosmetic formulations (emulsion, cleansing, and conditioner). Methods: Three different concentrations of PEA (0.3%, 1%, and 2.5%) were used in cosmetic formulations. The physical appearance of the formulas was assessed manually, and the antimicrobial nature of PEA and PEA-containing cosmetic formulations was evaluated by agar well plate assay. Results: The use of PEA has not affected the physical appearance and quality of the formulations, except the high concentration of PEA in the cleansing solution, which reduced the foam formation. The minimal required concentration of PEA in emulsions and cleansings was 1.0% and 2.5% in the conditioners. -

Conversion of Alcohols Into Amines by Borrowing Hydrogen
University of Bath PHD Conversion of Alcohols into Amines by Borrowing Hydrogen Hamid, Malai Award date: 2008 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 03. Oct. 2021 Conversion of Alcohols into Amines by Borrowing Hydrogen Malai Haniti Sheikh Abdul Hamid A thesis submitted for the degree of Doctor of Philosophy Department of Chemistry University of Bath November 2008 COPYRIGHT Attention is drawn to the fact that copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without the prior written consent of the author. -

Tin-Free Alternatives to the Barton-Mccombie Deoxygenation of Alcohols to Alkanes Involving Reductive Electron Transfer
The French connecTion CHIMIA 2016, 70, No. 1/2 67 doi:10.2533/chimia.2016.67 Chimia 70 (2016) 67–76 © Swiss Chemical Society Tin-free Alternatives to the Barton- McCombie Deoxygenation of Alcohols to Alkanes Involving Reductive Electron Transfer Ludwig Chenneberg and Cyril Ollivier* Abstract: Echoing the recent celebration of the fortieth anniversary of the Barton-McCombie reaction, this review aims to explore another facet of radical processes for deoxygenation of alcohols by considering SET (single electron transfer) reduction of carboxylic ester, thiocarbonate and thiocarbamate derivatives. Various protocols have been developed relying on the use of organic and organometallic SET reagents, electrochemi- cal conditions, photoinduced electron transfer processes and visible-light photoredox catalysis. Applications to the synthesis of molecules of interest provide a glimpse into the scope of these different approaches. Keywords: Alcohols · Deoxygenation · Electron transfer · Radicals · Reduction 1. Introduction phorus derivatives and less toxic metals.[1] secondary and tertiary alcohols.[7] In 2015, Interesting perspectives have opened up the radical community celebrated the 40th Radical chemistry has witnessed an ex- with the use of organoboranes[4] and com- anniversary of the discovery of the Barton- plosive growth over the last three decades. pounds responsible for electron-transfer McCombie deoxygenation reaction, unan- Owing to their mildness and high com- reactions have been increasingly seen as imously considered as the most common