The Female Urogynecological Microbiome: Determining Niche Specificity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BD™ Gardnerella Selective Agar with 5% Human Blood
INSTRUCTIONS FOR USE – READY-TO-USE PLATED MEDIA PA-254094.06 Rev.: July 2014 BD Gardnerella Selective Agar with 5% Human Blood INTENDED USE BD Gardnerella Selective Agar with 5% Human Blood is a partially selective and differential medium for the isolation of Gardnerella vaginalis from clinical specimens. PRINCIPLES AND EXPLANATION OF THE PROCEDURE Microbiological method. Gardnerella vaginalis is considered to be one of the organisms causing vaginitis.1-4 Although the organism may be present in a high percentage of normal women in the vaginal flora, its importance as a cause of non-specific vaginitis (also called bacterial vaginosis) has never been questioned. In symptomatic women, G. vaginalis frequently is associated with anaerobes such as Prevotella bivia, P. disiens, Mobiluncus, Peptostreptococcus, and/or others which are a regular part of the urethral or intestinal, but not vaginal flora. In non-specific vaginitis, normal Lactobacillus flora is reduced or absent. Gardnerella vaginalis is considered to be the indicator organism for non-specific vaginitis which, in fact, is a polymicrobial infection.3,4 Although non- culture methods such as a direct Gram stain have been recommended in recent years for genital specimens, culture is still preferred by many laboratories.1,5 G. vaginalis may also be responsible for a variety of other diseases such as preterm birth, chorioamnionitis, urinary tract infections, newborn infections, and septicemia.6 The detection of the organism on routinely used media is difficult since Gardnerella and other -

Product Sheet Info
Product Information Sheet for HM-289 Facklamia sp., Strain HGF4 Incubation: Temperature: 37°C Catalog No. HM-289 Atmosphere: Aerobic Propagation: 1. Keep vial frozen until ready for use, then thaw. For research use only. Not for human use. 2. Transfer the entire thawed aliquot into a single tube of broth. Contributor: 3. Use several drops of the suspension to inoculate an Thomas M. Schmidt, Professor, Department of Microbiology agar slant and/or plate. and Molecular Genetics, Michigan State University, East 4. Incubate the tube, slant and/or plate at 37°C for 48 to Lansing, Michigan, USA 168 hours. Manufacturer: Citation: BEI Resources Acknowledgment for publications should read “The following reagent was obtained through BEI Resources, NIAID, NIH as Product Description: part of the Human Microbiome Project: Facklamia sp., Strain Bacteria Classification: Aerococcaceae, Facklamia HGF4, HM-289.” Species: Facklamia sp. Strain: HGF4 Biosafety Level: 2 Original Source: Facklamia sp., strain HGF4 is a human Appropriate safety procedures should always be used with gastrointestinal isolate.1 this material. Laboratory safety is discussed in the following Comments: Facklamia sp., strain HGF4 (HMP ID 9411) is a publication: U.S. Department of Health and Human Services, reference genome for The Human Microbiome Project Public Health Service, Centers for Disease Control and (HMP). HMP is an initiative to identify and characterize Prevention, and National Institutes of Health. Biosafety in human microbial flora. The complete genome of Facklamia Microbiological and Biomedical Laboratories. 5th ed. sp., strain HGF4 is currently being sequenced at the J. Washington, DC: U.S. Government Printing Office, 2009; see Craig Venter Institute. -

Bacterial Profiles and Antibiogrants of the Bacteria Isolated of the Exposed Pulps of Dog and Cheetah Canine Teeth
Bacterial profiles and antibiogrants of the bacteria isolated of the exposed pulps of dog and cheetah canine teeth A dissertation submitted to the Faculty of Veterinary Science, University of Pretoria. In partial fulfillment of the requirements for the degree Master of Science (Veterinary Science) Promoter: Dr. Gerhard Steenkamp Co-promoter: Ms. Anna-Mari Bosman Department of Companion Animal Clinical Studies Faculty of Veterinary Science University of Pretoria Pretoria (January 2012) J.C. Almansa Ruiz © University of Pretoria Declaration I declare that the dissertation that I hereby submit for the Masters of Science degree in Veterinary Science at the University of Pretoria has not previously been submitted by me for degree purposes at any other university. J.C. Almansa Ruiz ii Dedications To one of the most amazing hunters of the African bush, the Cheetah, that has made me dream since I was a child, and the closest I had been to one, before starting this project was in National Geographic documentaries. I wish all of them a better future in which their habitat will be more respected. To all conservationists, especially to Carla Conradie and Dave Houghton, for spending their lives saving these animals which are suffering from the consequences of the encroachment of human beings into their territory. Some, such as George Adamson, the lion conservationist, even lost their lives in this mission. To the conservationist, Lawrence Anthony, for risking his life in a suicide mission to save the animals in the Baghdad Zoo, when the conflict in Iraq exploded. To my mother and father Jose Maria and Rosa. -

Multi-Product Lactic Acid Bacteria Fermentations: a Review
fermentation Review Multi-Product Lactic Acid Bacteria Fermentations: A Review José Aníbal Mora-Villalobos 1 ,Jéssica Montero-Zamora 1, Natalia Barboza 2,3, Carolina Rojas-Garbanzo 3, Jessie Usaga 3, Mauricio Redondo-Solano 4, Linda Schroedter 5, Agata Olszewska-Widdrat 5 and José Pablo López-Gómez 5,* 1 National Center for Biotechnological Innovations of Costa Rica (CENIBiot), National Center of High Technology (CeNAT), San Jose 1174-1200, Costa Rica; [email protected] (J.A.M.-V.); [email protected] (J.M.-Z.) 2 Food Technology Department, University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] 3 National Center for Food Science and Technology (CITA), University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] (C.R.-G.); [email protected] (J.U.) 4 Research Center in Tropical Diseases (CIET) and Food Microbiology Section, Microbiology Faculty, University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] 5 Bioengineering Department, Leibniz Institute for Agricultural Engineering and Bioeconomy (ATB), 14469 Potsdam, Germany; [email protected] (L.S.); [email protected] (A.O.-W.) * Correspondence: [email protected]; Tel.: +49-(0331)-5699-857 Received: 15 December 2019; Accepted: 4 February 2020; Published: 10 February 2020 Abstract: Industrial biotechnology is a continuously expanding field focused on the application of microorganisms to produce chemicals using renewable sources as substrates. Currently, an increasing interest in new versatile processes, able to utilize a variety of substrates to obtain diverse products, can be observed. -

Human Microbiota Network: Unveiling Potential Crosstalk Between the Different Microbiota Ecosystems and Their Role in Health and Disease
nutrients Review Human Microbiota Network: Unveiling Potential Crosstalk between the Different Microbiota Ecosystems and Their Role in Health and Disease Jose E. Martínez †, Augusto Vargas † , Tania Pérez-Sánchez , Ignacio J. Encío , Miriam Cabello-Olmo * and Miguel Barajas * Biochemistry Area, Department of Health Science, Public University of Navarre, 31008 Pamplona, Spain; [email protected] (J.E.M.); [email protected] (A.V.); [email protected] (T.P.-S.); [email protected] (I.J.E.) * Correspondence: [email protected] (M.C.-O.); [email protected] (M.B.) † These authors contributed equally to this work. Abstract: The human body is host to a large number of microorganisms which conform the human microbiota, that is known to play an important role in health and disease. Although most of the microorganisms that coexist with us are located in the gut, microbial cells present in other locations (like skin, respiratory tract, genitourinary tract, and the vaginal zone in women) also play a significant role regulating host health. The fact that there are different kinds of microbiota in different body areas does not mean they are independent. It is plausible that connection exist, and different studies have shown that the microbiota present in different zones of the human body has the capability of communicating through secondary metabolites. In this sense, dysbiosis in one body compartment Citation: Martínez, J.E.; Vargas, A.; may negatively affect distal areas and contribute to the development of diseases. Accordingly, it Pérez-Sánchez, T.; Encío, I.J.; could be hypothesized that the whole set of microbial cells that inhabit the human body form a Cabello-Olmo, M.; Barajas, M. -

First Report of Human Intracranial Weeksella Virosa Infection in the Setting of Anaplastic Meningiomata
Case report Title of paper: First report of human intracranial Weeksella virosa infection in the setting of anaplastic meningiomata Authors: Sebastian M Toescu1 Sandra Lacey2 Hu Liang Low1 Affiliations: 1. Department of Neurosurgery, Essex Neurosciences Centre, Queens Hospital, Romford, RM7 0AG, UK 2. Department of Microbiology, Queens Hospital, Romford, RM7 0AG, UK Corresponding author: Sebastian M Toescu, Department of Neurosurgery, Essex Neurosciences Centre, Queens Hospital, Romford, RM7 0AG, UK, [email protected] Abstract: A 49 year old female underwent multiple craniotomies for resection of aggressive meningeal tumours (WHO Grade III). She re-presented to hospital with sepsis due to ventriculitis. The craniotomy wound was urgently debrided and isolates of the Gram negative rod Weeksella virosa identified on 16S PCR. This species is most commonly found as a genitourinary commensal and here we report the first documented case of intracranial infection with this species and its successful treatment with a 6-week course of oral -lactam antibiotics. Keywords: Weeksella virosa; central nervous system infection; ventriculitis; anaplastic meningioma 1 Clinical details A 49-year old, right handed female presented with early morning headaches and vomiting. Her past medical history was unremarkable. On examination, the only focal deficit elicited was a dense left homonymous hemianopia. Magnetic resonance imaging (MRI) of the brain revealed a lesion occupying the trigone and occipital horn of the right lateral ventricle (Figure 1A). No other intracranial lesions were seen, and computed tomography scans of the chest, abdomen and pelvis did not show any abnormalities. A complete macroscopic excision of the tumour was effected without problems. The postoperative MRI scan did not show any residual enhancing tumour tissue. -
JET Microbiological Efficacy Summary
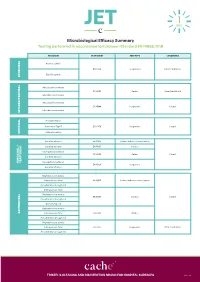
Microbiological Efficacy Summary Testing performed in accordance to European Standard EN 14885:2018 ORGANISM TEST NORM TEST TYPE CONDITIONS Bacillus subtilis EN 13704 Suspension Clean 1 and Dirty 1 Bacillus cereus SPORICIDAL Mycobacterium terrae EN 14563 Carrier Clean 1 and Dirty 2 Mycobacterium avium Mycobacterium terrae EN 14348 Suspension Clean 1 Mycobacterium avium MYCOBACTERICIDAL Poliovirus Type 1 Adenovirus Type 5 EN 14476 Suspension Clean 1 Murine Norovirus VIRUCIDAL Candida albicans EN 16615 Surface with mechanical action Candida albicans EN 13697 Surface Aspergillus brasiliensis EN 14562 Carrier Clean 1 Candida albicans YEASTICIDAL FUNGICIDAL / FUNGICIDAL Aspergillus brasiliensis EN 13624 Suspension Candida albicans Staphylococcus aureus Enterococcus hirae EN 16615 Surface with mechanical action Pseudomonas aeruginosa Enterococcus hirae Staphylococcus aureus EN 13697 Surface Clean 1 Pseudomonas aeruginosa Escherichia coli Staphylococcus aureus BACTERICIDAL Enterococcus hirae EN 14561 Carrier Pseudomonas aeruginosa Staphylococcus aureus Enterococcus hirae EN 13727 Suspension Clean 1 and Dirty 1 Pseudomonas aeruginosa TRISTEL’S CLEANING AND DISINFECTION BRAND FOR HOSPITAL SURFACES Page 1 of 3 Additional Testing TEST METHOD RNA DNA / Polyacrylamide gel electrophoresis (PAGE) ORGANISM TEST METHOD TEST TYPE CONDITIONS Acanthamoeba castellanii cysts Following the method of EN 13704 Suspension Clean 1 PROTOZOA Bacillus subtilis EN 17126 Suspension Clean 1 Bacillus cereus Clostridium difficile EN 13704 Suspension Clean 1 and Dirty 1 -

Biodegradation Treatment of Petrochemical Wastewaters
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS DEPARTAMENTO DE BIOLOGIA VEGETAL Biodegradation treatment of petrochemical wastewaters Catarina Isabel Nunes Alexandre Dissertação Mestrado em Microbiologia Aplicada Orientadores Doutora Sandra Sanches Professora Doutora Lélia Chambel 2015 Biodegradation treatment of petrochemical wastewaters Catarina Isabel Nunes Alexandre 2015 This thesis was fully performed at the Institute of Experimental and Technologic Biology (IBET) of Instituto de Tecnologia Química e Bioquímica (ITQB) under the direct supervision of Drª Sandra Sanches in the scope of the Master in Applied Microbiology of the Faculty of Sciences of the University of Lisbon. Prof. Drª Lélia Chambel was the internal designated supervisor in the scope of the Master in Applied Microbiology of the Faculty of Sciences of the University of Lisbon. Agradecimentos Gostaria de agradecer a todas as pessoas que estiveram directamente ou indirectamente envolvidas na execução da minha tese de mestrado, pois sem eles a sua realização não teria sido possível. Queria começar por agradecer à Doutora Sandra Sanches, que se demonstrou sempre disponível para esclarecer dúvidas quando precisei, e que fez questão de me ensinar de forma rigorosa e exigente. À Doutora Maria Teresa Crespo, que assim que lhe pedi para me orientar me disse que sim imediatamente, fez questão de me treinar em vários contextos e sempre estimulou o meu envolvimento nas rotinas do laboratório. À Professora Doutora Lélia Chambel, que sempre me esclareceu dúvidas sobre processos burocráticos, me deu conselhos quando eu mais precisei e que me apoiou durante toda a minha tese. Queria também agradecer à Doutora Dulce Brito, que sempre se mostrou disponível para ajudar quando a nossa equipa mais precisava dela, e sempre me ajudou a realizar as tarefas mais básicas do meu trabalho até eu ter ganho a minha autonomia no laboratório. -

NOTES in Vitro Activities of Norfloxacin and Ciprofloxacin Against
ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, July 1984, p. 94-96 Vol. 26, No. 1 0066-4804/84/070094-03$02.00/0 Copyright C 1984, American Society for Microbiology NOTES In Vitro Activities of Norfloxacin and Ciprofloxacin Against Mycobacterium tuberculosis, M. avium Complex, M. chelonei, M. fortuitum, and M. kansasii J. DOUGLAS GAY, DONALD R. DEYOUNG, AND GLENN D. ROBERTS* Section of Clinical Microbiology, Department of Laboratory Medicine, Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55905 Received 28 November 1983/Accepted 4 April 1984 The activities of ciprofloxacin and norfloxacin against 100 mycobacteria isolates were studied in vitro by the 1% standard proportion method. Ciprofloxacin was more active against M. tuberculosis and M. fortuitum with MICs of 1.0 and 0.25 ,ug/ml, respectively, against 90% of isolates; norfloxacin had MICs of 8.0 and 2.0 ,ug/ml, respectively, against 90% of isolates. Nalidixic acid and other heterocyclic carbonic acid deriva- studied. The organisms were taken from the Mayo Clinic tives have been used primarily in the treatment of urinary stock culture collection, which included recent clinical iso- tract infections for many years. The compounds of this lates. Stock cultures were maintained on Middlebrook 7H10 general group include nalidixic acid, oxolinic acid, pipemidic agar slants (Difco Laboratories, Detroit, Mich.) and were acid, cinoxacin, and rosoxacin. Two new substances in this subcultured monthly. The identification of isolates was series which have been recently synthesized are norfloxacin based on standard biochemical tests (17) and gas-liquid (6) (1-ethyl-6-fluoro-1,4-dihydro-4-oxo-7-[ 1-piperazinyl ]-3- chromatography (16). -

Bacterial Diversity and Functional Analysis of Severe Early Childhood
www.nature.com/scientificreports OPEN Bacterial diversity and functional analysis of severe early childhood caries and recurrence in India Balakrishnan Kalpana1,3, Puniethaa Prabhu3, Ashaq Hussain Bhat3, Arunsaikiran Senthilkumar3, Raj Pranap Arun1, Sharath Asokan4, Sachin S. Gunthe2 & Rama S. Verma1,5* Dental caries is the most prevalent oral disease afecting nearly 70% of children in India and elsewhere. Micro-ecological niche based acidifcation due to dysbiosis in oral microbiome are crucial for caries onset and progression. Here we report the tooth bacteriome diversity compared in Indian children with caries free (CF), severe early childhood caries (SC) and recurrent caries (RC). High quality V3–V4 amplicon sequencing revealed that SC exhibited high bacterial diversity with unique combination and interrelationship. Gracillibacteria_GN02 and TM7 were unique in CF and SC respectively, while Bacteroidetes, Fusobacteria were signifcantly high in RC. Interestingly, we found Streptococcus oralis subsp. tigurinus clade 071 in all groups with signifcant abundance in SC and RC. Positive correlation between low and high abundant bacteria as well as with TCS, PTS and ABC transporters were seen from co-occurrence network analysis. This could lead to persistence of SC niche resulting in RC. Comparative in vitro assessment of bioflm formation showed that the standard culture of S. oralis and its phylogenetically similar clinical isolates showed profound bioflm formation and augmented the growth and enhanced bioflm formation in S. mutans in both dual and multispecies cultures. Interaction among more than 700 species of microbiota under diferent micro-ecological niches of the human oral cavity1,2 acts as a primary defense against various pathogens. Tis has been observed to play a signifcant role in child’s oral and general health. -

A Genomic Journey Through a Genus of Large DNA Viruses
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Virology Papers Virology, Nebraska Center for 2013 Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses Adrien Jeanniard Aix-Marseille Université David D. Dunigan University of Nebraska-Lincoln, [email protected] James Gurnon University of Nebraska-Lincoln, [email protected] Irina V. Agarkova University of Nebraska-Lincoln, [email protected] Ming Kang University of Nebraska-Lincoln, [email protected] See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/virologypub Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, Cell and Developmental Biology Commons, Genetics and Genomics Commons, Infectious Disease Commons, Medical Immunology Commons, Medical Pathology Commons, and the Virology Commons Jeanniard, Adrien; Dunigan, David D.; Gurnon, James; Agarkova, Irina V.; Kang, Ming; Vitek, Jason; Duncan, Garry; McClung, O William; Larsen, Megan; Claverie, Jean-Michel; Van Etten, James L.; and Blanc, Guillaume, "Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses" (2013). Virology Papers. 245. https://digitalcommons.unl.edu/virologypub/245 This Article is brought to you for free and open access by the Virology, Nebraska Center for at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Virology Papers by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Adrien Jeanniard, David D. Dunigan, James Gurnon, Irina V. Agarkova, Ming Kang, Jason Vitek, Garry Duncan, O William McClung, Megan Larsen, Jean-Michel Claverie, James L. Van Etten, and Guillaume Blanc This article is available at DigitalCommons@University of Nebraska - Lincoln: https://digitalcommons.unl.edu/ virologypub/245 Jeanniard, Dunigan, Gurnon, Agarkova, Kang, Vitek, Duncan, McClung, Larsen, Claverie, Van Etten & Blanc in BMC Genomics (2013) 14. -

Influence of Host Plants on the Diversity of Gut Microbiota
Inuence of Host Plants on the Diversity of Gut Microbiota Communities of Fall Armyworm Spodoptera frugiperda J.E. Smith (Lepidoptera: Noctuidae) Juliana Amaka Ugwu ( [email protected] ) Forestry Research Institute of Nigeria https://orcid.org/0000-0003-1862-6864 Fred Asiegbu Department of Forest sciences, University of Helsinki, P.O Box 27, FIN-00014 Helsinki, Finland. Research Article Keywords: host plants, microbial variability, fall armyworm, larvae, bacteria Posted Date: June 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-657579/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract The gut bacteria of insects inuence their host physiology positively, although their mechanism is not yet understood. This study characterized the microbiome of the gut of Spodoptera frugiperda larvae fed with nine different host plants; sugar cane (M1), maize (M2), onion (M3), cucumber (R1), tomato (R2), sweet potato (R3), cabbage L1), green amaranth (L2), and celocia (L3) by sequencing the theV3-V4 hypervariable region of the 16S rRNA gene using Illumina PE250 NovaSeq system. The results revealed that gut bacterial composition varied among larvae samples fed on different host plants. Three alpha diversity indices revealed highly signicant differences on the gut bacterial diversity of S. frugiperda fed with different host plants.. Analysis of Molecular Variance (AMOVA) and Analysis of Similarity (ANOSIM) also revealed signicant variations on the bacterial communities among the various host plants. Five bacteria phyla (Firmicutes, Proteobacteria, Cyanobacteria, Actinobacteria and Bacteroidetes) were prevalent across the larvae samples. Firmicutes (44.1%) was the most dominant phylum followed by Proteobacteria (28.5%).