Approach to Anemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hemolysis and Venous Thrombosis: Which Link? A
ISSN: 2378-3656 Rkiouak et al. Clin Med Rev Case Rep 2020, 7:329 DOI: 10.23937/2378-3656/1410329 Volume 7 | Issue 11 Clinical Medical Reviews Open Access and Case Reports ORIGINAL RESEARCH Hemolysis and Venous Thrombosis: Which Link? A. Rkiouak, PH.D1, I El Kassimi, MD2, N Sahel, MD2, M Zaizaa, MD2 and Y Sekkach, PhD1 Check for updates Internal Medicine A Department, Mohammed V Military Hospital Medical School of Rabat, Morocco *Corresponding author: Adil Rkiouak, PH.D., Department of Internal Medicine A, Mohammed V Military Hospital, Medical School of Rabat, Morocco, Tel: +212-66-179-44-04 The mechanism of antibody-mediated hemolysis is Abstract via phagocytosis or complement-mediated destruction The association hemolysis and venous thrombosis remains and can occur intravascular or extravascular. The intra- unknown to clinicians, despite our advances in comrehen- sion of pathophysiological bases. vascular mechanisms include direct cellular destruction via lysis, toxins, or trauma; fragmentation and oxida- Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow’s triad. It is not sur- tion. prising that there is a link between haemolytic disorders and Multiple haemolytic disorders produce substantial thrombosis. intravascular haemolysis. Examples, the corpuscular We will try to clarify the main pro-thrombotic mechanisms hemolysis include PNH, extra-corpuscular haemolysis, during hemolysis through 3 clinical observations of deep ve- acquired (autoimmune haemolytic anaemia (AIHA , nous thrombosis in 3 main types of hemolytic pathologies, ) namely a case of paroxysmal nocturnal hemoglobinuria, thrombotic thrombocytopenic purpura (PTT)), as well thrombotic thrombocytopenic purpura and autoimmune as other diseases. These disorders are also associated anemia hemolytic. -

Clostridial Septicemia with Intravascular Hemolysis: a Case Report G
Henry Ford Hospital Medical Journal Volume 13 | Number 4 Article 4 12-1965 Clostridial Septicemia With Intravascular Hemolysis: A Case Report G. M. Mastio E. Morfin Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons Recommended Citation Mastio, G. M. and Morfin, E. (1965) "Clostridial Septicemia With Intravascular Hemolysis: A Case Report," Henry Ford Hospital Medical Bulletin : Vol. 13 : No. 4 , 421-425. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol13/iss4/4 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Bull. Vol. 13, December 1965 CLOSTRIDIAL SEPTICEMIA WITH INTRAVASCULAR HEMOLYSIS A CASE REPORT G. M. MASTIC, M.D. AND E. MORFIN, M.D. In 1871 Bottini' demonstrated the bacterial nature of gas gangrene, but failed to isolate a causal organism. Clostridium perfringens, sometimes known as Clostridium welchii, was discovered independently during 1892 and 1893 by Welch, Frankel, "Veillon and Zuber.^ This organism is a saprophytic inhabitant of the intestinal tract, and may be a harmless saprophyte of the female genital tract occurring in the vagina in 4-6 per cent of pregnant women. Clostridial organisms occur in great numbers and distribution throughout the world. Because of this, they are very common in traumatic wounds. Very few species of Clostridia, however, are pathogenic, and still fewer are capable of producing gas gangrene in man. -

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Viral Hepatitis with Acute Hemoglobinuria
22 Case Report Viral Hepatitis with Acute Hemoglobinuria Jagabandhu Ghosh1 Joydeep Ghosh2 1 Department of Paediatrics, I.P.G.M.E.R and S.S.K.M Hospital, Kolkata, Address for correspondence Jagabandhu Ghosh, MD (PAED), Ushashi West Bengal, India Housing Society, 245 Vivekananda Road, Kolkata 700006, India 2 Department of Biotechnology, Heritage Institute of Technology, (e-mail: [email protected]). Kolkata, West Bengal, India J Pediatr Infect Dis 2015;10:22–24. Abstract A 7-year-old male child presented with moderate degree fever, yellowish discoloration of eyes and urine. Examination on admission revealed severe anemia, jaundice, hepato- Keywords megaly, and splenomegaly. On the day following admission, the child showed evidence ► hepatitis of blackish discoloration of urine. The diagnosis was established as viral A hepatitis with ► G6PD glucose-6-phosphate dehydrogenase (G6PD) deficiency. The child recovered with ► viral supportive therapy. We suggest that either universal immunization against hepatitis ► hemolysis A, or routine newborn screening for G6PD deficiency, could prevent the serious ► intravascular morbidity or mortality that can occur when these two conditions coexist. Introduction of eyes, and urine for the past 5 days before admission. There was no history of hematemesis, melena, or any other bleed- Acute viral hepatitis A is widely prevalent in India.1 Viral ing, swelling of body, convulsion, and drug ingestion just hepatitis is the leading of acute hepatitis in children in our before present illness or any previous blood transfusion. His country, and hepatitis A is the most common type of viral urine volume was satisfactory. On examination, the child was hepatitis. Hepatitis A does not commonly present with severe deeply icteric, severely anemic, and drowsy. -
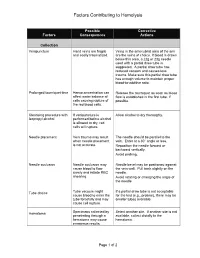
Factors Contributing to Hemolysis
Factors Contributing to Hemolysis Possible Corrective Factors Consequences Actions Collection Venipuncture Hand veins are fragile Veins in the antecubital area of the arm and easily traumatized. are the veins of choice. If blood is drawn below this area, a 22g or 23g needle used with a partial draw tube is suggested. A partial draw tube has reduced vacuum and causes less trauma. Make sure this partial draw tube has enough volume to maintain proper blood-to-additive ratio. Prolonged tourniquet time Hemoconcentration can Release the tourniquet as soon as blood affect water balance of flow is established in the first tube, if cells causing rupture of possible. the red blood cells. Cleansing procedure with If venipuncture is Allow alcohol to dry thoroughly. isopropyl alcohol performed before alcohol is allowed to dry, red cells will rupture. Needle placement Vein trauma may result The needle should be parallel to the when needle placement vein. Enter at a 30° angle or less. is not accurate. Reposition the needle forward or backward vertically. Avoid probing. Needle occlusion Needle occlusion may Needle bevel may be positioned against cause blood to flow the vein wall. Pull back slightly on the slowly and initiate RBC needle. shearing. Avoid rotating or changing the angle of the needle. Tube choice Tube vacuum might If a partial draw tube is not acceptable cause blood to enter the for the test (e.g., protime), there may be tube forcefully and may smaller tubes available. cause cell rupture. Hematoma Specimens collected by Select another site. If another site is not penetrating through a available, collect distally to the hematoma may cause hematoma. -

PATHOLOGY RESIDENT HEMATOLOGY ROTATION (North Florida/South Georgia Veterans Health Care System): Rotation Director: William L
PATHOLOGY RESIDENT HEMATOLOGY ROTATION (North Florida/South Georgia Veterans Health Care System): Rotation Director: William L. Clapp, M.D., Chief, Hematology Section, Gainesville VAMC; Consultants: Neil S. Harris, M.D., Director, Laboratory Hematology/Coagulation, University of Florida and Shands Hospital and Raul C. Braylan, M.D., Director, Hematopathology, University of Florida and Shands Hospital 1. Description of the Rotation: In this rotation, the resident will gain experience in laboratory hematology, which will include (1) peripheral blood studies to evaluate a variety of hematologic disorders, including anemias, lymphoproliferative and myeloproliferative disorders and leukemias. The emphasis on a multidisciplinary approach to diagnose hematologic disorders (including correlation of the peripheral blood studies with bone marrow and lymph node studies) provides an opportunity for the resident to also gain additional experience in (2) traditional histopathology, (3) immunohistochemistry, (4) electron microscopy, (5) protein electrophoresis, (6) flow cytometry, (7) cytogenetics and (8) molecular genetics which may be performed on the peripheral blood, bone marrow or lymph nodes of patients. The residents will acquire valuable experience by independently performing some bone marrow procedures. In addition, the resident will gain experience in coagulation testing. The residents will become familiar with the instrumentation in the hematology laboratory, including the operating principles and trouble-shooting (medical knowledge). The availability of assembled case study sets and reading materials (medical knowledge) will enhance the resident’s experience. Participation in CAP surveys, continuing education and hematology conferences is a component of the rotation (practice-based learning). Management issues and computer applications will be discussed (practice-based learning). As appropriate to the individual case or consultation under review, the ethical, socioeconomic, medicolegal and cost-containment issues will be reviewed and discussed. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

Hematopoiesis/RBC Disorders
Neonatal Hematopoiesis and RBC Disorders Vandy Black, MD, MSc Pediatric Hematology June 2, 2016 Objectives • Review normal erytHropoiesis in tHe fetus and neonate and regulation of fetal hemoglobin • Outline tHe differential diagnosis of neonatal RBC disorders witH a focus on tHe clinical and laboratory findings • Discuss common presentations of intrinsic red cell disorders in neonates WhicH of tHe following infants is most likely to be diagnosed with a primary hematologic disorder (i.e. need ongoing follow-up in my office)? A. A full-term male with a Hb of 7.5 gm/dL at birth (MCV 108) B. A one week old witH a newborn screen tHat shows Hb FAS C. A full-term Caucasian male with a peak bilirubin of 21 mg/dL whose mom is AB+ D. A 26 week AA female wHose fatHer Has a history of G6PD deficiency RBC Disorders in tHe NICU • Anemia is a common finding in tHe NICU • Differential is broad • Hospitalized preterm infants receive more PRBC transfusions tHan any otHer patient group • >80% of ELBW infants receive at least one PRBC transfusion How RBC Disorders Present? • Anemia on a CBC – May be an expected or incidental finding • Abnormal RBC indices • Abnormal newborn screens • Hyperbilirubinemia • Screening because of family History What is Normal? CHristensen et al, Semin Perinatol 2009 What is Normal? CHristensen et al, Semin Perinatol 2009 Hemoglobin SwitcHing How to ApproacH Anemia • Are otHer cell lines involved? • What is tHe MCV? • What is tHe reticulocyte count? • What does tHe peripHeral blood smear sHow? Microcytic Anemia • Iron Deficiency – Iron supplementation for preterm infants • Thalassemia – Beta-thalassemia less likely in the neonatal period • Chronic Inflammation – Disorders of iron transport (e.g. -

Acquired Hemophilia A: Pathogenesis and Treatment
Bleeding disorders Acquired hemophilia A: pathogenesis and treatment P.W. Collins ABSTRACT Arthur Bloom Haemophilia Centre, Acquired hemophilia A is an autoimmune disease caused by an inhibitory antibody to factor VIII. The School of Medicine, severity of bleeding varies but patients remain at risk of life-threatening bleeding until the inhibitor Cardiff University, Heath Park, has been eradicated. The cornerstones of management are rapid and accurate diagnosis, control of Cardiff, UK bleeding, investigation for an underlying cause, and eradication of the inhibitor by immunosuppres - sion. Patients should be managed jointly with a specialist center even if they present without signifi - cant bleeding. Despite an extensive literature, few controlled data are available and management Hematology Education: guidelines are based on expert opinion. Recombinant factor VIIa and activated prothrombin complex the education program for the concentrate are equally efficacious for treating bleeds and both are superior to factor VIII or desmo - annual congress of the European pressin. Immunosuppression should be started as soon as the diagnosis is made. Commonly used reg - Hematology Association imens are steroids alone or combined with cytotoxic agents. Rituximab is being used more commonly but current evidence does not suggest that it improves outcomes or reduces side effects. 2012;6:65-72 Introduction Pathogenesis Acquired hemophilia A (AHA) is a bleed - AHA is associated with autoimmune dis - ing disorder caused by polyclonal IgG1 and eases, such as rheumatoid arthritis, polymyal - IgG4 autoantibodies to the factor VIII ( FVIII ) gia rheumatic, and systemic lupus erythe - A2 and C2 domain. Morbidity and mortality matosis; malignancy; pregnancy and dermato - are high secondary to age, underlying dis - logical disorders, such as pemphigoid. -

A Distant Gene Deletion Affects Beta-Globin Gene Function in an Atypical Gamma Delta Beta-Thalassemia
A distant gene deletion affects beta-globin gene function in an atypical gamma delta beta-thalassemia. P Curtin, … , A D Stephens, H Lehmann J Clin Invest. 1985;76(4):1554-1558. https://doi.org/10.1172/JCI112136. Research Article We describe an English family with an atypical gamma delta beta-thalassemia syndrome. Heterozygosity results in a beta-thalassemia phenotype with normal hemoglobin A2. However, unlike previously described cases, no history of neonatal hemolytic anemia requiring blood transfusion was obtained. Gene mapping showed a deletion that extended from the third exon of the G gamma-globin gene upstream for approximately 100 kilobases (kb). The A gamma-globin, psi beta-, delta-, and beta-globin genes in cis remained intact. The malfunction of the beta-globin gene on a chromosome in which the deletion is located 25 kb away suggests that chromatin structure and conformation are important for globin gene expression. Find the latest version: https://jci.me/112136/pdf A Distant Gene Deletion Affects ,8-Globin Gene Function in an Atypical '6y5-Thalassemia Peter Curtin, Mario Pirastu, and Yuet Wai Kan Howard Hughes Medical Institute and Department ofMedicine, University of California, San Francisco, California 94143 John Anderson Gobert-Jones Department ofPathology, West Suffolk County Hospital, Bury St. Edmunds IP33-2QZ, Suffolk, England Adrian David Stephens Department ofHaematology, St. Bartholomew's Hospital, London ECIA-7BE, England Herman Lehmann Department ofBiochemistry, University ofCambridge, Cambridge CB2-lQW, England Abstract tologic picture of f3-thalassemia minor in adult life. Globin syn- thetic studies reveal a ,3 to a ratio of -0.5, but unlike the usual We describe an English family with an atypical 'yS6-thalassemia fl-thalassemia heterozygote, the levels of HbA2 (and HbF) are syndrome. -

Anemia in Heart Failure - from Guidelines to Controversies and Challenges
52 Education Anemia in heart failure - from guidelines to controversies and challenges Oana Sîrbu1,*, Mariana Floria1,*, Petru Dascalita*, Alexandra Stoica1,*, Paula Adascalitei, Victorita Sorodoc1,*, Laurentiu Sorodoc1,* *Grigore T. Popa University of Medicine and Pharmacy; Iasi-Romania 1Sf. Spiridon Emergency Hospital; Iasi-Romania ABSTRACT Anemia associated with heart failure is a frequent condition, which may lead to heart function deterioration by the activation of neuro-hormonal mechanisms. Therefore, a vicious circle is present in the relationship of heart failure and anemia. The consequence is reflected upon the pa- tients’ survival, quality of life, and hospital readmissions. Anemia and iron deficiency should be correctly diagnosed and treated in patients with heart failure. The etiology is multifactorial but certainly not fully understood. There is data suggesting that the following factors can cause ane- mia alone or in combination: iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunc- tion. There is data suggesting the association among iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunction. The main pathophysiologic mechanisms, with the strongest evidence-based medicine data, are iron deficiency and inflammation. In clinical practice, the etiology of anemia needs thorough evaluation for determining the best possible therapeutic course. In this context, we must correctly treat the patients’ diseases; according with the current guidelines we have now only one intravenous iron drug. This paper is focused on data about anemia in heart failure, from prevalence to optimal treatment, controversies, and challenges. (Anatol J Cardiol 2018; 20: 52-9) Keywords: anemia, heart failure, intravenous iron, ferric carboxymaltose, quality of life Introduction g/dL in men) (2). -

Simultaneous Pure Red Cell Aplasia and Auto-Immune Hemolytic Anemia in a Previously Healthy Infant
Simultaneous Pure Red Cell Aplasia and Auto-immune Hemolytic Anemia in a Previously Healthy Infant Robert P. Sanders, MD July 16, 2002 Case Presentation Patient Z.H. • Previously Healthy 7 month old WM • Presented to local ER 6/30 with 1 wk of decreased activity and appetite, low grade temp, 2 day h/o pallor. • Noted to have severe anemia, transferred to LeBonheur • Review of Systems – ? Single episode of dark urine – 4 yo sister diagnosed with Fifth disease 1 wk prior to onset of symptoms, cousin later also diagnosed with Fifth disease – Otherwise negative ROS •PMH – Term, no complications – Normal Newborn Screen – Hospitalized 12/01 with RSV • Medications - None • Allergies - NKDA • FH - Both parents have Hepatitis C (pt negative) • SH - Lives with Mom, 4 yo sister • Development Normal Physical Exam • 37.2 167 33 84/19 9.3kg • Gen - Alert, pale, sl yellow skin tone, NAD •HEENT -No scleral icterus • CHEST - Clear • CV - RRR, II/VI SEM at LLSB • ABD - Soft, BS+, no HSM • SKIN - No Rash • NEURO - No Focal Deficits Labs •CBC – WBC 20,400 • 58% PMN 37% Lymph 4% Mono 1 % Eo – Hgb 3.4 • MCV 75 MCHC 38.0 MCH 28.4 – Platelets 409,000 • Retic 0.5% • Smear - Sl anisocytosis, Sl hypochromia, Mod microcytes, Sl toxic granulation • G6PD Assay 16.6 U/g Hb (nl 4.6-13.5) • DAT, Broad Spectrum Positive – IgG negative – C3b, C3d weakly positive • Chemistries – Total Bili 2.0 – Uric Acid 4.8 –LDH 949 • Urinalysis Negative, Urobilinogen 0.2 • Blood and Urine cultures negative What is your differential diagnosis? Differential Diagnosis • Transient Erythroblastopenia of Childhood • Diamond-Blackfan syndrome • Underlying red cell disorder with Parvovirus induced Transient Aplastic Crisis • Immunohemolytic anemia with reticulocytopenia Hospital Course • Admitted to ICU for observation, transferred to floor 7/1.