Revealing the Developmental Dynamics in Male Strobilus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Variation in Sex Expression in Canada Yew (Taxus Canadensis) Author(S): Taber D
Variation in Sex Expression in Canada Yew (Taxus canadensis) Author(s): Taber D. Allison Source: American Journal of Botany, Vol. 78, No. 4 (Apr., 1991), pp. 569-578 Published by: Botanical Society of America Stable URL: http://www.jstor.org/stable/2445266 . Accessed: 23/08/2011 15:56 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at . http://www.jstor.org/page/info/about/policies/terms.jsp JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Botanical Society of America is collaborating with JSTOR to digitize, preserve and extend access to American Journal of Botany. http://www.jstor.org AmericanJournal of Botany 78(4): 569-578. 1991. VARIATION IN SEX EXPRESSION IN CANADA YEW (TAXUS CANADENSIS)1 TABER D. ALLISON2 JamesFord Bell Museumof Natural History and Departmentof Ecology and BehavioralBiology, Universityof Minnesota, Minneapolis, Minnesota 55455 Sex expressionwas measuredin severalCanada yew (Taxus canadensisMarsh.) populations of theApostle Islands of Wisconsinand southeasternMinnesota to determinethe extent of variationwithin and among populations. Sex expression was recorded qualitatively (monoecious, male,or female) and quantitatively (by male to female strobilus ratios or standardized phenotypic gender).No discernibletrends in differencesin sex expressionamong populations or habitats wererecorded. Trends in sexexpression of individuals within populations were complex. Small yewstended to be maleor, if monoecious, had female-biasedstrobilus ratios. -

Earliest Record of Megaphylls and Leafy Structures, and Their Initial Diversification
Review Geology August 2013 Vol.58 No.23: 27842793 doi: 10.1007/s11434-013-5799-x Earliest record of megaphylls and leafy structures, and their initial diversification HAO ShouGang* & XUE JinZhuang Key Laboratory of Orogenic Belts and Crustal Evolution, School of Earth and Space Sciences, Peking University, Beijing 100871, China Received January 14, 2013; accepted February 26, 2013; published online April 10, 2013 Evolutionary changes in the structure of leaves have had far-reaching effects on the anatomy and physiology of vascular plants, resulting in morphological diversity and species expansion. People have long been interested in the question of the nature of the morphology of early leaves and how they were attained. At least five lineages of euphyllophytes can be recognized among the Early Devonian fossil plants (Pragian age, ca. 410 Ma ago) of South China. Their different leaf precursors or “branch-leaf com- plexes” are believed to foreshadow true megaphylls with different venation patterns and configurations, indicating that multiple origins of megaphylls had occurred by the Early Devonian, much earlier than has previously been recognized. In addition to megaphylls in euphyllophytes, the laminate leaf-like appendages (sporophylls or bracts) occurred independently in several dis- tantly related Early Devonian plant lineages, probably as a response to ecological factors such as high atmospheric CO2 concen- trations. This is a typical example of convergent evolution in early plants. Early Devonian, euphyllophyte, megaphyll, leaf-like appendage, branch-leaf complex Citation: Hao S G, Xue J Z. Earliest record of megaphylls and leafy structures, and their initial diversification. Chin Sci Bull, 2013, 58: 27842793, doi: 10.1007/s11434- 013-5799-x The origin and evolution of leaves in vascular plants was phology and evolutionary diversification of early leaves of one of the most important evolutionary events affecting the basal euphyllophytes remain enigmatic. -

X. the Conifers and Ginkgo
X. The Conifers and Ginkgo Now we turn our attention to the Coniferales, another great assemblage of seed plants. First let's compare the conifers with the cycads: Cycads Conifers few apical meristems per plant many apical meristems per plant leaves pinnately divided leaves undivided wood manoxylic wood pycnoxylic seeds borne on megaphylls seeds borne on stems We should also remember that these two groups have a lot in common. To begin with, they are both groups of woody seed plants. They are united by a small set of derived features: 1) the basic structure of the stele (a eustele or a sympodium, two words for the same thing) and no leaf gaps 2) the design of the apical meristem (many initials, subtended by a slowly dividing group of cells called the central mother zone) 3) the design of the tracheids (circular-bordered pits with a torus) We have three new seed plant orders to examine this week: A. Cordaitales This is yet another plant group from the coal forest. (Find it on the Peabody mural!) The best-known genus, Cordaites, is a tree with pycnoxylic wood bearing leaves up to about a foot and a half long and four inches wide. In addition, these trees bore sporangia (micro- and mega-) in strobili in the axils of these big leaves. The megasporangia were enclosed in ovules. Look at fossils of leaves and pollen-bearing shoots of Cordaites. The large, many-veined megaphylls are ancestral to modern pine needles; the shoots are ancestral to pollen-bearing strobili of modern conifers. 67 B. -

Seasonal Growth of the Female Strobilus in Pinus Radiata
No. 1 15 SEASONAL GROWTH OF THE FEMALE STROBILUS IN PINUS RADIATA G. B. SWEET and M. P. BOLLMANN Forest Research Institute, New Zealand Forest Service, Rotorua (Received for publication 12 November, 1970) ABSTRACT Growth of female strobili of Pinus radiata D. Don from central North Island of New Zealand is described and illustrated with photographs. The two-and-a-half year period from strobilus emergence until cone maturity comprises a seasonal period of growth in which pollination occurs, a second period of seasonal growth in which fertilisation occurs, and finally a period of cone maturation. The periods of rapid growth do not appear to result directly from either pollination or fertilisation, and the seasonal growth periods have some similarity to those of vegetative growth. The time taken to reach cone maturity in P. radiata (a closed-cone pine) is six months longer than that frequently described for other species of Pinus. INTRODUCTION The general pattern of female strobilus development in Pinus is well documented (e.g., Ferguson, 1904; Stanley, 1958; Sarvas, 1962). Broadly, a total period of two-and- a-half years is involved, leading through from strobilus determination one summer, to anthesis the following spring, to fertilisation late in the subsequent spring and finally to maturation the succeeding autumn. Seed production in Pinus radiata D. Don apparently follows within general limits the typical pattern for Pinus, but few details have been published either of its strobilus or its ovule development. As part of a comprehensive study of the processes leading to seed production in this species, material was collected in 1968 and 1969 to enable details of strobilus development to be determined. -

Multipurpose Trees for Agroforestry in the Pacific Islands
For Educators, Gardeners, Farmers, Foresters, and Landscapers Agroforestry Guides for Pacific Islands “Well-researched, concise, user-friendly...an invaluable practical resource for those working to conserve and expand the use of trees in agricultural systems.” —APANews, The Asia-Pacific Agroforestry Newsletter FAO Regional Office, Bangkok, Thailand “A significant contribution to public education, advancing the cause of integrated agriculture and forestry...a resource of lasting value.” —The Permaculture Activist, North Carolina “A most excellent handbook...a wonderful resource.” —Developing Countries Farm Radio Network, Toronto, Canada “Eloquently makes a case for reintroducing and emphasizing trees in our island agriculture.” —Dr. Bill Raynor, Program Director, The Nature Conservancy, Pohnpei, Federated States of Micronesia “Provides a real clearinghouse on traditional and modern agroforestry not only for Pacific Islands, also very useful for other regions.” —ILEIA Newsletter for Low External Input and Sustainable Agriculture, The Netherlands Purchase the book at http://www.agroforestry.net/afg/ Agroforestry Guides for Pacific Islands edited by Craig R. Elevitch and Kim M. Wilkinson Price: $24.95 (plus shipping) Availability: Usually ships within one business day. Paperback - 240 pages, illustrated and fully indexed Release date: September, 2000 ISBN: 0970254407 Publisher: Permanent Agriculture Resources, P.O. Box 428, Holualoa, HI, 96725, USA. Tel: 808-324-4427, Fax: 808-324-4129, email: [email protected] Agroforestry Guides for Pacific Islands #2 Multipurpose Trees for Agroforestry in the Pacific Islands by Randolph R. Thaman, Craig R. Elevitch, and Kim M. Wilkinson www.agroforestry.net Multipurpose Trees for Agroforestry in the Pacific Islands Abstract: The protection and planting of trees in agroforestry systems can serve as an important, locally achievable, and cost-effective step in sustainable development in the Pacific Islands. -

Wild Food Plants of Remote Oceania
Acta Societatis Botanicorum Poloniae Journal homepage: pbsociety.org.pl/journals/index.php/asbp INVITED REVIEW Received: 2012.09.01 Accepted: 2012.10.12 Published electronically: 2012.11.22 Acta Soc Bot Pol 81(4):371–380 DOI: 10.5586/asbp.2012.034 Wild food plants of Remote Oceania Will C. McClatchey* Botanical Research Institute of Texas, 1700 University Drive, Fort Worth, Texas 76107, USA Department of Botany, University of Hawai’i at Manoa, Honolulu, Hawai’i 96822, USA Environmental Studies Program, Texas Christian University, Fort Worth, Texas 76107, USA Abstract Agricultural societies partly depend upon wild foods. Relationships between an agricultural society and its wild foods can be explored by examining how the society responds through colonization of new lands that have not been previously inhabited. The oldest clear example of this phenomenon took place about 5000 years ago in the tropical Western Pacific at the “boundary” interface between Near and Remote Oceania. An inventory of wild and domesticated food plants used by people living along “the remote side of” that interface has been prepared from the literature. This was then assessed for the roles of plants at the time of original colonization of Remote Oceania. The majority of species are wild foods, and most of these are used as leafy vegetables and fruits. The wild food plants mostly serve as supplements to domesticated species, although there are a few that can be used as substitutes for traditional staples. Keywords: Oceania, wild food plants, colonization, agriculture Introduction implication is that humans brought wild plant materials to their homes or processing sites, much as squirrels or fruit bats Human-plant co-evolutionary relationships have been might do, and then consumed less than all of the material col- documented for processes of wild plant food domestication lected, discarding material (seeds or other living parts) that into socially critical crops [1,2]. -

1 Relationships of Angiosperms To
Relationships of Angiosperms to 1 Other Seed Plants Seed plants are of fundamental importance both evolution- all gymnosperms (living and extinct) together are not arily and ecologically. They dominate terrestrial landscapes, monophyletic. Importantly, several fossil lineages, Cayto- and the seed has played a central role in agriculture and hu- niales, Bennettitales, Pentoxylales, and Glossopteridales man history. There are fi ve extant lineages of seed plants: (glossopterids), have been proposed as putative close rela- angiosperms, cycads, conifers, gnetophytes, and Ginkgo. tives of the angiosperms based on phylogenetic analyses These fi ve groups have usually been treated as distinct (e.g., Crane 1985; Rothwell and Serbet 1994; reviewed in phyla — Magnoliophyta (or Anthophyta), Cycadophyta, Doyle 2006, 2008, 2012; Friis et al. 2011). These fossil lin- Co ni fe ro phyta, Gnetophyta, and Ginkgophyta, respec- eages, sometimes referred to as the para-angiophytes, will tively. Cantino et al. (2007) used the following “rank- free” therefore be covered in more detail later in this chapter. An- names (see Chapter 12): Angiospermae, Cycadophyta, other fossil lineage, the corystosperms, has been proposed Coniferae, Gnetophyta, and Ginkgo. Of these, the angio- as a possible angiosperm ancestor as part of the “mostly sperms are by far the most diverse, with ~14,000 genera male hypothesis” (Frohlich and Parker 2000), but as re- and perhaps as many as 350,000 (The Plant List 2010) to viewed here, corystosperms usually do not appear as close 400,000 (Govaerts 2001) species. The conifers, with ap- angiosperm relatives in phylogenetic trees. proximately 70 genera and nearly 600 species, are the sec- The seed plants represent an ancient radiation, with ond largest group of living seed plants. -

Evolutionary Studies of the Gnetales
Evolutionary studies of the Gnetales Chen Hou Academic dissertation for the degree of Doctor of Philosophy in Plant Sys- tematics presented at Stockholm University 2016 Evolutionary studies of the Gnetales Chen Hou ©Chen Hou Stockholm University 2016 ISBN 978-91-7649-371-7 Printed in Sweden by US AB, Stockholm 2016 Distributor: Department of Ecology, Environment and Plant Sciences, Stockholm University, Sweden ”Although relatively few people have chosen to study the Gnetales, those who have had the opportunity to work with these organisms experience a profound sense of extraordinary beauty and complexity of the evolutionary process.” - Willian E. Friedman, 1996 Abstract The Gnetales consist of three distinct genera, Ephedra, Gnetum and Wel- witschia with considerable divergence among them regarding morphologi- cal, ecological and molecular characters. A longstanding debate of the simi- larity between the Gnetales and angiosperms and the unresolved seed plant phylogeny intrigues plant scientists to further investigate the evolutionary history of the Gnetales. The presented projects deal with interdisciplinary questions on proteomics, chloroplast genomes, phylogenetic relationships, gross morphology and taxonomy. The thesis aims to summarize general problems encountered in previous studies, and to provide new insights and future perspectives based on the results of completed and ongoing projects. In Ephedra, the Mediterranean species E. foeminea has been shown to be entomophilous and it possesses an important phylogenetic status as the sister of the remaining genus. Therefore, the chloroplast genome of E. foeminea was assembled and compared to that previously presented (of the anemophi- lous Asian species E. equisetina, nested in the core clade of Ephedra). The genome has a quadripartite structure and comprises 118 genes and 109,584 base pairs. -

BAGO Gnetum Gnemon Linn
RESEARCH INFORMATION SERIES ON ECOSYSTEMS Volume 13 No. 2 May – August 2001 BAGO Gnetum gnemon Linn. Compiled by Rafael T. Cadiz Helen B. Florido Foreword In search of locally available natural resources that could be a productive source of livelihood for farmers, we came up with an alternative tree species called Bago (Gnetum gnemon L.). Bago is a multi purpose tree species which is native to the Philippines. It can be a source of food, wood and fiber. Because of its edible young shoot, the species is more popularly known in some upland areas as a vegetable crop. Unknown to our local farmers, Bago trees can become a source of export products. Countries like Indonesia process the seeds and leaves of Bago and export them to Japan and some European countries. Aside from its economic potential, bago can also help in rehabilitating our marginal lands. This species aids in soil improvement because of its beneficial association with some mychorrhizal fungi. Both an economically and ecologically important tree species, Bago could provide great benefits to our rural communities. The information contained in this issue will serve as guide to our readers especially the upland farmers in considering bago for agroforestry, tree planting activities, or tree plantation development. CELSO P. DIAZ Director 1. Common name: Bago 2. Local names: bago, magatungal (Lanao, Cotabato); bago or bagu (Bataan, Tayabas, Camarines); banago (Visaya, Bohol); kunan (Davao); nabo (Bicol) 3. Internationally used common name: Spanish koint fir (English) Other common names: Melinjo, belinjo, bagoe - Indonesia Maninjau - Malaysia Voe, Khalet - Cambodia Peedae, phak, miang phak kaniang, liang - Thailand Gam cay, bet - Vietnam 4. -

122 Antibacterial Effect of Gnetum Gnemon L. Leaves
Online-ISSN 2565-1409 Journal of Widya Medika Junior Vol 3. No. 2 April 2021 ANTIBACTERIAL EFFECT OF GNETUM GNEMON L. LEAVES EXTRACT ON STAPHYLOCOCCUS AUREUS Paul J Dayoh1), Endang Isbandiati2), Titien Rahayu3) ABSTRACT Introduction: Bacterial infections are common among doctors in Indonesia, leading to increased use of antibiotics. Increased use of antibiotics, if not accompanied by rational indication, can increase bacterial resistance. At present, alternative medicine from natural ingredients is widely studied, one of which is the leaves of melinjo (Gnetum gnemon L.). Melinjo leaves are thought to contain antibacterial compounds such as flavonoids, tannins, saponins, and alkaloids. Staphylococcus aureus is a round Gram-positive bacteria, that forms grape-like clusters. Staphylococcus aureus is a normal flora on the skin and human mucosa but can be opportunistic pathogens that cause mild to severe infections such as sepsis. Pathogenic Staphylococcus aureus can produce hemolysis blood, frozen plasma, and produce various extracellular enzymes and toxins. Purpose: This research aims to study the MIC and MBC of melinjo leaves extract on the growth of Staphylococcus aureus bacteria. This research is an experimental study with a post- test control group design. Method: Testing conducted using the microdilution method. Melinjo leaves used were extracted by maceration method using 96% ethanol. Samples were Staphylococcus aureus ATCC 25923 from BBLK Surabaya. Results: extract of melinjo leaves (Gnetum gnemon L.) had MIC on Staphylococcus aureus bacteria at a concentration of 640 mg/ml. MBC was not obtained because the extract of melinjo leaves clotted above 1280 mg/ml concentration. Conclusion: Melinjo leaves extract has an antibacterial effect against Staphylococcus aureus, the MIC is obtained at a concentration of 640 mg/ml, and the MBC cannot be determined. -
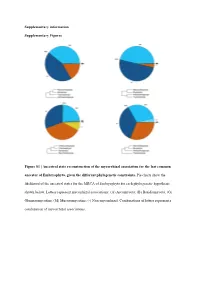
Ancestral State Reconstruction of the Mycorrhizal Association for the Last Common Ancestor of Embryophyta, Given the Different Phylogenetic Constraints
Supplementary information Supplementary Figures Figure S1 | Ancestral state reconstruction of the mycorrhizal association for the last common ancestor of Embryophyta, given the different phylogenetic constraints. Pie charts show the likelihood of the ancestral states for the MRCA of Embryophyta for each phylogenetic hypothesis shown below. Letters represent mycorrhizal associations: (A) Ascomycota; (B) Basidiomycota; (G) Glomeromycotina; (M) Mucoromycotina; (-) Non-mycorrhizal. Combinations of letters represent a combination of mycorrhizal associations. Austrocedrus chilensis Chamaecyparis obtusa Sequoiadendron giganteum Prumnopitys taxifolia Prumnopitys Prumnopitys montana Prumnopitys Prumnopitys ferruginea Prumnopitys Araucaria angustifolia Araucaria Dacrycarpus dacrydioides Dacrycarpus Taxus baccata Podocarpus oleifolius Podocarpus Afrocarpus falcatus Afrocarpus Ephedra fragilis Nymphaea alba Nymphaea Gnetum gnemon Abies alba Abies balsamea Austrobaileya scandens Austrobaileya Abies nordmanniana Thalictrum minus Thalictrum Abies homolepis Caltha palustris Caltha Abies magnifica ia repens Ranunculus Abies religiosa Ranunculus montanus Ranunculus Clematis vitalba Clematis Keteleeria davidiana Anemone patens Anemone Tsuga canadensis Vitis vinifera Vitis Tsuga mertensiana Saxifraga oppositifolia Saxifraga Larix decidua Hypericum maculatum Hypericum Larix gmelinii Phyllanthus calycinus Phyllanthus Larix kaempferi Hieronyma oblonga Hieronyma Pseudotsuga menziesii Salix reinii Salix Picea abies Salix polaris Salix Picea crassifolia Salix herbacea -

Endangered Plants and Their Uses of Sivasagar District, Assam, India
International Research Journal of Biological Sciences ___________________________________ ISSN 2278-3202 Vol. 4(7), 15-18, July (2015) Int. Res. J. Biological Sci. Endangered Plants and their uses of Sivasagar District, Assam, India P Jiji Department of Botany, Swahid Peoli Phukan College, Namti Namtidole, Sivasagar, Assam, INDIA Available online at: www.isca.in, www.isca.me Received 10 th May 2015, revised 13 th June 2015, accepted 3rd July 2015 Abstract Assam is a strategically very important state of North Eastern region. It has a treasure of biodiversity. Sivasagar District of Assam is a historical place was capital city of ahom dynasty. The district is also important due to their natural resources. The district have six reserve forest areas, viz. Sapekati, Dilli, Abhaypur, Sola, Gelekey, and Panidihing reserve forests, having treasure of biodiversity due to wide variability in climatic and edaphic condition. Major parts of the flora of Assam are found in Sivasagar District. Ethnic people (Tai-Ahom, Tai-Shyam, Tai-Phake, Mising, Kachari, Sonowal and various Tea-tribe) are living neighboring these forests. They depend the forest for their shelter, food, medicines, fodder etc. But biodiversity of district is declining fast and most of the plants were coming under IUCN red list due to increased demand, destructive harvesting over exploitation, illegal trade, opens grazing, developing buildings etc. To meet the demand of authentic plant materials from the field, plantation of medicinal plant farm helps for sustainable utilization. Keywords : Endangered, ethno medicinal plants, conservation, sustainable use. Introduction Material and Methods Assam is the gateway of Northeastern India, having an area of The study based on fieldwork, literary survey, herbarium 78,438 km 2 and is one of the most sensitive biodiversity zones scrutiny of plant specimen.