Predictive Model for Inflammation Grades of Chronic Hepatitis B: Large-Scale Analysis of Clinical Parameters and Gene Expressions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
DEPs in osteosarcoma cells comparing to osteoblastic cells Biological Process Protein Percentage of Hits metabolic process (GO:0008152) 29.3 29.3% cellular process (GO:0009987) 20.2 20.2% localization (GO:0051179) 9.4 9.4% biological regulation (GO:0065007) 8 8.0% developmental process (GO:0032502) 7.8 7.8% response to stimulus (GO:0050896) 5.6 5.6% cellular component organization (GO:0071840) 5.6 5.6% multicellular organismal process (GO:0032501) 4.4 4.4% immune system process (GO:0002376) 4.2 4.2% biological adhesion (GO:0022610) 2.7 2.7% apoptotic process (GO:0006915) 1.6 1.6% reproduction (GO:0000003) 0.8 0.8% locomotion (GO:0040011) 0.4 0.4% cell killing (GO:0001906) 0.1 0.1% 100.1% Genes 2179Hits 3870 biological adhesion apoptotic process … reproduction (GO:0000003) , 0.8% (GO:0022610) , 2.7% locomotion (GO:0040011) ,… immune system process cell killing (GO:0001906) , 0.1% (GO:0002376) , 4.2% multicellular organismal process (GO:0032501) , metabolic process 4.4% (GO:0008152) , 29.3% cellular component organization (GO:0071840) , 5.6% response to stimulus (GO:0050896), 5.6% developmental process (GO:0032502) , 7.8% biological regulation (GO:0065007) , 8.0% cellular process (GO:0009987) , 20.2% localization (GO:0051179) , 9. -

ADVISORY COMMISSION on CHILDHOOD VACCINES TABLE of CONTENTS December 8, 2017
ADVISORY COMMISSION ON CHILDHOOD VACCINES TABLE OF CONTENTS December 8, 2017 TAB • ACCV Agenda 1 • ACCV Charter • ACCV Roster • 2017 Meeting Dates • Meeting Minutes 2 o Draft Minutes – September 8, 2017 • Vaccine Injury Compensation Trust Fund Statement 3 o Vaccine Injury Compensation Trust Fund Summary Sheet for the Period of 10/1/2016 – 9/30/2017 • VICP Data and Statistics 4 • Meeting Presentations & Updates 5 o Report from the Division of Injury Compensation Programs 5.1 o Report from the Department of Justice 5.2 o Petitions to Add Injuries to the Vaccine Injury Table Introduction 5.3 o Petition to Add Tics as an Injury to the Vaccine Injury Table 5.4 o Petition to Add Asthma as an Injury to the Vaccine Injury Table 5.5 5.6 o Petition to Add Pediatric Autoimmune Neuropsychiatric Syndrome (PANS), Pediatric Infection-Triggered Autoimmune Neuropsychiatric Disorders (PITANDS), and Pediatric Autoimmune Neuropsychiatric Disorders (PANDAS) as Injuries to the Vaccine Injury Table o Petition to Add Experimental Autoimmune Encephalomyelitis (EAE) and/or 5.7 Acute Demyelinating Encephalomyelitis (ADEM) as injuries to the Vaccine Injury Table 5.8 o Update on the Immunization Safety Office Vaccine Activities (CDC) o Update on the National Institute of Allergy and Infectious Diseases Vaccine 5.9 Activities (NIH) o Update on the Center for Biologics, Evaluation and Research Vaccine 5.10 Activities (FDA) 5.11 o Update from the National Vaccine Program Office • Program Related Articles 6 6.1 o Popular Science, “Why Are We So Bad At Producing The Right -

Malonylation of GAPDH Is an Inflammatory Signal in Macrophages
ARTICLE https://doi.org/10.1038/s41467-018-08187-6 OPEN Malonylation of GAPDH is an inflammatory signal in macrophages Silvia Galván-Peña1,2, Richard G. Carroll3, Carla Newman4, Elizabeth C. Hinchy5, Eva Palsson-McDermott1, Elektra K. Robinson6, Sergio Covarrubias 6, Alan Nadin7, Andrew M. James5, Moritz Haneklaus1, Susan Carpenter6, Vincent P. Kelly 1, Michael P. Murphy 5, Louise K. Modis2 & Luke A. O’Neill1,2 Macrophages undergo metabolic changes during activation that are coupled to functional 1234567890():,; responses. The gram negative bacterial product lipopolysaccharide (LPS) is especially potent at driving metabolic reprogramming, enhancing glycolysis and altering the Krebs cycle. Here we describe a role for the citrate-derived metabolite malonyl-CoA in the effect of LPS in macrophages. Malonylation of a wide variety of proteins occurs in response to LPS. We focused on one of these, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). In resting macrophages, GAPDH binds to and suppresses translation of several inflammatory mRNAs, including that encoding TNFα. Upon LPS stimulation, GAPDH undergoes malonylation on lysine 213, leading to its dissociation from TNFα mRNA, promoting translation. We therefore identify for the first time malonylation as a signal, regulating GAPDH mRNA binding to promote inflammation. 1 School of Biochemistry and Immunology, Trinity Biomedical Science Institute, Trinity College, Dublin D2, Ireland. 2 Immunology Catalyst, GlaxoSmithKline, Stevenage SG1 2NY, UK. 3 Department of Molecular and Cellular Therapeutics, Royal College of Surgeons in Ireland, Dublin D2, Ireland. 4 In Vitro/In Vivo Translation, GlaxoSmithKline, Stevenage SG1 2NY, UK. 5 MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge CB2 0XY, UK. 6 Department of Molecular Cell and Developmental Biology, UC Santa Cruz, Santa Cruz 95064 CA, USA. -

Kidney V-Atpase-Rich Cell Proteome Database
A comprehensive list of the proteins that are expressed in V-ATPase-rich cells harvested from the kidneys based on the isolation by enzymatic digestion and fluorescence-activated cell sorting (FACS) from transgenic B1-EGFP mice, which express EGFP under the control of the promoter of the V-ATPase-B1 subunit. In these mice, type A and B intercalated cells and connecting segment principal cells of the kidney express EGFP. The protein identification was performed by LC-MS/MS using an LTQ tandem mass spectrometer (Thermo Fisher Scientific). For questions or comments please contact Sylvie Breton ([email protected]) or Mark A. Knepper ([email protected]). -

Loss of the E3 Ubiquitin Ligase MKRN1 Represses Diet-Induced Metabolic Syndrome Through AMPK Activation
ARTICLE DOI: 10.1038/s41467-018-05721-4 OPEN Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation Min-Sik Lee1, Hyun-Ji Han2, Su Yeon Han2, Il Young Kim3,4, Sehyun Chae5, Choong-Sil Lee2, Sung Eun Kim2, Seul Gi Yoon4, Jun-Won Park4, Jung-Hoon Kim2, Soyeon Shin2, Manhyung Jeong2, Aram Ko2, Ho-Young Lee6, Kyoung-Jin Oh 7, Yun-Hee Lee 8, Kwang-Hee Bae7, Seung-Hoi Koo9, Jea-woo Kim10, Je Kyung Seong3,4, Daehee Hwang5 & Jaewhan Song 2 1234567890():,; AMP-activated protein kinase (AMPK) plays a key role in controlling energy metabolism in response to physiological and nutritional status. Although AMPK activation has been pro- posed as a promising molecular target for treating obesity and its related comorbidities, the use of pharmacological AMPK activators has been met with contradictory therapeutic challenges. Here we show a regulatory mechanism for AMPK through its ubiquitination and degradation by the E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1). MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation due to AMPK stabilisation and activation. Accordingly, MKRN1-null mice show chronic AMPK activation in both liver and adipose tissue, resulting in significant suppression of diet-induced metabolic syndrome. We demonstrate also its therapeutic effect by administering shRNA targeting MKRN1 into obese mice that reverses non-alcoholic fatty liver disease. We suggest that ubiquitin-dependent AMPK degradation represents a target therapeutic strategy for meta- bolic disorders. 1 Harvard Medical School, Boston Children’s Hospital, 3 Blackfan Circle CLS-16060.2, Boston, MA 02115, USA. 2 Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Republic of Korea. -

Detection of H3k4me3 Identifies Neurohiv Signatures, Genomic
viruses Article Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis Liana Basova 1, Alexander Lindsey 1, Anne Marie McGovern 1, Ronald J. Ellis 2 and Maria Cecilia Garibaldi Marcondes 1,* 1 San Diego Biomedical Research Institute, San Diego, CA 92121, USA; [email protected] (L.B.); [email protected] (A.L.); [email protected] (A.M.M.) 2 Departments of Neurosciences and Psychiatry, University of California San Diego, San Diego, CA 92103, USA; [email protected] * Correspondence: [email protected] Abstract: Human postmortem specimens are extremely valuable resources for investigating trans- lational hypotheses. Tissue repositories collect clinically assessed specimens from people with and without HIV, including age, viral load, treatments, substance use patterns and cognitive functions. One challenge is the limited number of specimens suitable for transcriptional studies, mainly due to poor RNA quality resulting from long postmortem intervals. We hypothesized that epigenomic Citation: Basova, L.; Lindsey, A.; signatures would be more stable than RNA for assessing global changes associated with outcomes McGovern, A.M.; Ellis, R.J.; of interest. We found that H3K27Ac or RNA Polymerase (Pol) were not consistently detected by Marcondes, M.C.G. Detection of H3K4me3 Identifies NeuroHIV Chromatin Immunoprecipitation (ChIP), while the enhancer H3K4me3 histone modification was Signatures, Genomic Effects of abundant and stable up to the 72 h postmortem. We tested our ability to use H3K4me3 in human Methamphetamine and Addiction prefrontal cortex from HIV+ individuals meeting criteria for methamphetamine use disorder or not Pathways in Postmortem HIV+ Brain (Meth +/−) which exhibited poor RNA quality and were not suitable for transcriptional profiling. -
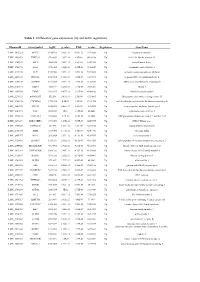
Table 1. Differential Gene Expression (Up and Down Regulation)
Table 1. Differential gene expression (up and down regulation). IlluminaId GeneSymbol logFC p value FDR t value Regulation GeneName ILMN_1814221 NPTX1 8120.166 2.02E-16 9.56E-12 107.0626 Up neuronal pentraxin 1 ILMN_1693471 KLHL35 879.4869 2.03E-14 4.8E-10 66.98158 Up kelch like family member 35 ILMN_1705153 NEFH 3866.938 3.55E-14 5.6E-10 63.27518 Up neurofilament heavy ILMN_1708778 ASS1 3751.105 6.81E-14 6.69E-10 59.20647 Up argininosuccinate synthase 1 ILMN_2114720 SLPI 1949.583 9.25E-14 7.29E-10 57.39296 Up secretory leukocyte peptidase inhibitor ILMN_2415583 HS6ST2 150.4964 1.51E-13 1.02E-09 54.59292 Up heparansulfate 6-O-sulfotransferase 2 ILMN_3185198 ACTR3C 112.0705 2.58E-13 1.53E-09 51.68897 Up ARP3 actin-related protein 3 homolog C ILMN_1700541 FBLN1 3126.79 3.36E-13 1.77E-09 50.31287 Up fibulin 1 ILMN_2109708 TYMP 922.6535 4.87E-13 2.31E-09 48.44836 Up thymidine phosphorylase ILMN_2214473 ARHGEF35 555.516 5.87E-13 2.5E-09 47.53665 Up Rho guanine nucleotide exchange factor 35 ILMN_1740170 CHCHD10 1770.965 6.4E-13 2.5E-09 47.11754 Up coiled-coil-helix-coiled-coil-helix domain containing 10 ILMN_1800739 SPINT2 1656.074 6.86E-13 2.5E-09 46.78296 Up serine peptidase inhibitor, Kunitz type 2 ILMN_3241522 ASS1 603.9339 1E-12 3.15E-09 45.0058 Up argininosuccinate synthase 1 ILMN_1740233 UGT1A10 540.0026 1.1E-12 3.15E-09 44.5868 Up UDP glucuronosyltransferase family 1 member A10 ILMN_3273229 MIR193BHG 273.3476 1.13E-12 3.15E-09 44.45755 Up MIR193B host gene ILMN_1748827 TMEM238 614.575 1.65E-12 3.8E-09 42.78856 Up transmembrane protein -

Impact of Exome Sequencing in Characterizing Mendelian Disorders
Journal of Human Genetics (2012) 57, 621–632 & 2012 The Japan Society of Human Genetics All rights reserved 1434-5161/12 $32.00 www.nature.com/jhg REVIEW Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders Bahareh Rabbani1, Nejat Mahdieh1, Kazuyoshi Hosomichi, Hirofumi Nakaoka and Ituro Inoue Traditional approaches for gene mapping from candidate gene studies to positional cloning strategies have been applied for Mendelian disorders. Since 2005, next-generation sequencing (NGS) technologies are improving as rapid, high-throughput and cost-effective approaches to fulfill medical sciences and research demands. Using NGS, the underlying causative genes are directly distinguished via a systematic filtering, in which the identified gene variants are checked for novelty and functionality. During the past 2 years, the role of more than 100 genes has been distinguished in rare Mendelian disorders by means of whole-exome sequencing (WES). Combination of WES with traditional approaches, consistent with linkage analysis, has had the greatest impact on those disorders following autosomal mode of inheritance; in more than 60 identified genes, the causal variants have been transmitted at homozygous or compound heterozygous state. Recent literatures focusing on identified new causal genes in Mendelian disorders using WES are reviewed in the present survey. Journal of Human Genetics (2012) 57, 621–632; doi:10.1038/jhg.2012.91; published online 26 July 2012 Keywords: exome sequencing; mendelian disorder; mutation; next-generation sequencing; NGS; WES INTRODUCTION genes and 4535 disorders with known molecular basis (http:// One of the major concerns of medical sciences is finding the causal omim.org/statistics/geneMap). -

Post-Translational Lysine Ac (Et) Ylation in Health, Ageing and Disease
Biol. Chem. 2021; ▪▪▪(▪▪▪): 1–44 Review Anna-Theresa Blasl, Sabrina Schulze, Chuan Qin, Leonie G. Graf, Robert Vogt and Michael Lammers* Post-translational lysine ac(et)ylation in health, ageing and disease https://doi.org/10.1515/hsz-2021-0139 protein function, how it is regulated enzymatically and non- Received February 8, 2021; accepted June 18, 2021; enzymatically, how a dysfunction in this post-translational published online August 23, 2021 machinery contributes to disease development. A focus is set on sirtuins and lysine acyltransferases as these are direct Abstract: The acetylation/acylation (ac(et)ylation) of lysine sensors and mediators of the cellular metabolic state. side chains is a dynamic post-translational modification Finally, this review highlights technological advances to (PTM) regulating fundamental cellular processes with im- study lysine ac(et)ylation. plications on the organisms’ ageing process: metabolism, transcription, translation, cell proliferation, regulation of Keywords: ageing; KDAC; longevity; lysine acetylation; the cytoskeleton and DNA damage repair. First identified to lysine acetyltransferases; lysine deacetylases; sirtuins; occur on histones, later studies revealed the presence of synthetic biology. lysine ac(et)ylation in organisms of all kingdoms of life, in proteins covering all essential cellular processes. A remarkable finding showed that the NAD+-dependent Introduction sirtuin deacetylase Sir2 has an impact on replicative lifespan in Saccharomyces cerevisiae suggesting that lysine acetyla- The human genome encodes approximately 20,000–25,000 tion has a direct role in the ageing process. Later studies proteins (International Human Genome Sequencing fi fi identi ed sirtuins as mediators for bene cial effects of 2004). The diversity of this proteome can be substantially ’ caloric/dietary restriction on the organisms health- or life- enlarged by processes such as alternative splicing and span. -

The Journal of the International Federation of Clinical Chemistry and Laboratory Medicine in This Issue
February 2017 ISSN 1650-3414 Volume 28 Number 1 Communications and Publications Division (CPD) of the IFCC Editor-in-chief : Prof. Gábor L. Kovács, MD, PhD, DSc Department of Laboratory Medicine, Faculty of Medicine, University of Pecs, Hungary e-mail: [email protected] The Journal of the International Federation of Clinical Chemistry and Laboratory Medicine In this issue Foreword of the editor Gábor L. Kovács 3 Recent advances in pediatric laboratory medicine Edgard Delvin 4 Pediatric obesity and cardiometabolic disorders: risk factors and biomarkers E. Levy, A.K. Saenger, M.W. Steffes, E. Delvin 6 Pediatric Metabolic Syndrome: pathophysiology and laboratory assessment Victoria Higgins, Khosrow Adeli 25 Pediatric reference intervals for biochemical markers H. Tahmasebi, V. Higgins, A. Fung, D. Truong, N. White-Al Habeeb, K. Adeli 43 The utility of CSF for the diagnosis of primary and secondary monoamine neurotransmitter deficiencies A.B. Burlina, A. Celato, G. Polo, C. Edini, A.P. Burlina 64 Pediatric reference intervals for transferrin saturation in the CALIPER cohort Victoria Higgins, Man Khun Chan, Khosrow Adeli 77 Congenital hyperinsulinism caused by a de novo mutation in the ABCC8 gene – a case report Z. Molnár, L. Balogh, J. Kappelmayer, L. Madar, É. Gombos, I. Balogh 85 Letter: Hemochromatosis, erythrocytosis and the JAK2 p.V617F mutation Stephen E. Langabeer 92 In this issue: Recent Advances in Pediatric Laboratory Medicine Foreword of the editor Editor in Chief: Gábor L. Kovács, M.D., Ph.D., DSc Professor Edgard Delvin (Canada) was asked for the Montreal Children’s Hospital, affiliated to be the guest-editor of the special issue on with McGill University until 2014. -

Dynamic Expression Profile, Regulatory Mechanism And
www.nature.com/scientificreports OPEN Dynamic Expression Profle, Regulatory Mechanism and Correlation with Egg-laying Received: 20 February 2018 Accepted: 21 May 2018 Performance of ACSF Gene Family Published: xx xx xxxx in Chicken (Gallus gallus) Weihua Tian1, Hang Zheng1, Liyu Yang1, Hong Li1,2,3, Yadong Tian1,2,3, Yanbin Wang1,2,3, Shijie Lyu4, Gudrun A. Brockmann4, Xiangtao Kang1,2,3 & Xiaojun Liu1,2,3 Acyl-CoA synthetases (ACSs) are responsible for acyl-CoA synthesis from nonpolar hydrophilic fatty acids and play a vital role in many metabolic processes. As a category of ACS isozymes, members of ACS family (ACSF1-3) participate in lipid metabolism; however, their expression patterns, regulatory mechanisms and efects on egg-laying performance in chicken are poorly understood. Our in vivo and in vitro studies showed that ACSF1-3 genes were extensively expressed, and their expression levels changed dynamically in the liver among diferent development stages. Moreover, ACSF1 expression was upregulated and ACSF2 expression was downregulated by estrogen, but ACSF3 showed no response to estrogen treatment. The regulatory efect of estrogen on ACSF1 expression was mediated via ERα. The ACSF2 was highly expressed in the liver in peak-laying hens compared with pre-laying and late-laying hens, and also highly expressed in the liver continued egg-laying hens compared with inactive egg- laying hens. It is suggested that hepatic ACSF2 expression level might relate to egg-laying performance in chicken. In conclusion, the expression of ACSF1 was upregulated by estrogen via ERα, and the expression of ACSF2 was downregulated by estrogen and might be related to egg-laying performance in chicken. -

Bioinformatic Analysis of Leishmania Donovani Long-Chain Fatty Acid-Coa Ligase As a Novel Drug Target
SAGE-Hindawi Access to Research Molecular Biology International Volume 2011, Article ID 278051, 14 pages doi:10.4061/2011/278051 Research Article Bioinformatic Analysis of Leishmania donovani Long-Chain Fatty Acid-CoA Ligase as a Novel Drug Target Jaspreet Kaur, Rameshwar Tiwari, Arun Kumar, and Neeloo Singh Drug Target Discovery & Development Division, Central Drug Research Institute (CSIR), Chattar Manzil Palace, Lucknow 226001, India Correspondence should be addressed to Neeloo Singh, [email protected] Received 14 January 2011; Revised 29 March 2011; Accepted 13 April 2011 Academic Editor: Hemanta K. Majumder Copyright © 2011 Jaspreet Kaur et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Fatty acyl-CoA synthetase (fatty acid: CoA ligase, AMP-forming; (EC 6.2.1.3)) catalyzes the formation of fatty acyl-CoA by a two-step process that proceeds through the hydrolysis of pyrophosphate. Fatty acyl-CoA represents bioactive compounds that are involved in protein transport, enzyme activation, protein acylation, cell signaling, and transcriptional control in addition to serving as substrates for beta oxidation and phospholipid biosynthesis. Fatty acyl-CoA synthetase occupies a pivotal role in cellular homeostasis, particularly in lipid metabolism. Our interest in fatty acyl-CoA synthetase stems from the identification of this enzyme, long-chain fatty acyl-CoA ligase (LCFA) by microarray analysis. We found this enzyme to be differentially expressed by Leishmania donovani amastigotes resistant to antimonial treatment. In the present study, we confirm the presence of long-chain fatty acyl-CoA ligase gene in the genome of clinical isolates of Leishmania donovani collected from the disease endemic area in India.