Protein Secondary Structure Prediction Based on Data Partition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Effective Computational Method Incorporating Multiple Secondary
Old Dominion University ODU Digital Commons Computer Science Faculty Publications Computer Science 2017 An Effective Computational Method Incorporating Multiple Secondary Structure Predictions in Topology Determination for Cryo-EM Images Abhishek Biswas Old Dominion University Desh Ranjan Old Dominion University Mohammad Zubair Old Dominion University Stephanie Zeil Old Dominion University Kamal Al Nasr See next page for additional authors Follow this and additional works at: https://digitalcommons.odu.edu/computerscience_fac_pubs Part of the Biochemistry Commons, Computer Sciences Commons, Molecular Biology Commons, and the Statistics and Probability Commons Repository Citation Biswas, Abhishek; Ranjan, Desh; Zubair, Mohammad; Zeil, Stephanie; Al Nasr, Kamal; and He, Jing, "An Effective Computational Method Incorporating Multiple Secondary Structure Predictions in Topology Determination for Cryo-EM Images" (2017). Computer Science Faculty Publications. 81. https://digitalcommons.odu.edu/computerscience_fac_pubs/81 Original Publication Citation Biswas, A., Ranjan, D., Zubair, M., Zeil, S., Al Nasr, K., & He, J. (2017). An effective computational method incorporating multiple secondary structure predictions in topology determination for cryo-em images. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 14(3), 578-586. doi:10.1109/tcbb.2016.2543721 This Article is brought to you for free and open access by the Computer Science at ODU Digital Commons. It has been accepted for inclusion in Computer Science Faculty Publications by an authorized administrator of ODU Digital Commons. For more information, please contact [email protected]. Authors Abhishek Biswas, Desh Ranjan, Mohammad Zubair, Stephanie Zeil, Kamal Al Nasr, and Jing He This article is available at ODU Digital Commons: https://digitalcommons.odu.edu/computerscience_fac_pubs/81 HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author IEEE/ACM Manuscript Author Trans Comput Manuscript Author Biol Bioinform. -

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Increasing the Accuracy of Single Sequence Prediction Methods Using a Deep Semi-Supervised Learning Framework Lewis Moffat 1,∗ and David T
Structural Bioinformatics Downloaded from https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab491/6313164 by guest on 07 July 2021 Increasing the Accuracy of Single Sequence Prediction Methods Using a Deep Semi-Supervised Learning Framework Lewis Moffat 1,∗ and David T. Jones 1,∗ 1Department of Computer Science, University College London, Gower Street, London WC1E 6BT, UK; and Francis Crick Institute, 1 Midland Road, London NW1 1AT, UK ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Motivation: Over the past 50 years, our ability to model protein sequences with evolutionary information has progressed in leaps and bounds. However, even with the latest deep learning methods, the modelling of a critically important class of proteins, single orphan sequences, remains unsolved. Results: By taking a bioinformatics approach to semi-supervised machine learning, we develop Profile Augmentation of Single Sequences (PASS), a simple but powerful framework for building accurate single- sequence methods. To demonstrate the effectiveness of PASS we apply it to the mature field of secondary structure prediction. In doing so we develop S4PRED, the successor to the open-source PSIPRED-Single method, which achieves an unprecedented Q3 score of 75.3% on the standard CB513 test. PASS provides a blueprint for the development of a new generation of predictive methods, advancing our ability to model individual protein sequences. Availability: The S4PRED model is available as open source software on the PSIPRED GitHub repository (https://github.com/psipred/s4pred), along with documentation. It will also be provided as a part of the PSIPRED web service (http://bioinf.cs.ucl.ac.uk/psipred/) Contact: [email protected], [email protected] Supplementary information: Supplementary data are available at Bioinformatics online. -

Packing of Secondary Structures II
7.88 Lecture Notes - 5 7.24/7.88J/5.48J The Protein Folding and Human Disease Packing of Secondary Structures • Packing of Helices against sheets • Packing of sheets against sheets • Parallel • Orthogonal Table: “Amino Acid Composition of the Ten Proteins and of the Residues at the Helix to Helix Interfaces” Name Total % Total At contacts % at contacts Gly 182 9 15 4 Ala 191 9 49 12 Val 151 7 46 12 Leu 148 7 48 12 Ile 114 6 36 9 Pro 67 3 41 1 Phe 68 3 25 6 Tyr 87 6 14 4 Trp 35 2 7 2 His 45 2 18 5 ½Cys 21 1 3 1 Met 29 1 10 3 Ser 165 8 19 5 Thr 132 7 21 5 Asp 112 6 14 4 Asn 113 6 13 3 Glu 94 5 13 3 Gln 70 3 12 3 Lys 125 6 19 5 Arg 76 4 13 3 A. Factors Contributing to Stability of Correctly Folded Native State 1. Major source of stability = removal of hydrophobic side chains atoms from the solvent and burying in environment which excludes the solvent (Entropic contribution from water structure). 1 2. Formation of hydrogen bonds between buried amide and carbonyl groups is maximized 3. Retention of backbone conformations close to the minimal energies. 4. Close packing means optimal Van der Waals interactions. You have read about alpha/beta proteins in Brandon and Tooze. B. Helix to Sheet Packing Lets examine buried contacts between the helices and the sheets. First a quick review of beta sheet structure: Colored transparency: Theoretical model, not actual sheet. -

Amino Acid Preference Against Beta Sheet Through Allowing Backbone Hydration Enabled by the Presence of Cation
Amino acid preference against beta sheet through allowing backbone hydration enabled by the presence of cation John N. Sharley, University of Adelaide. arXiv 2016-10-03 [email protected] Table of Contents 1 Abstract 1 2 Introduction 2 2.1 Alpha helix preferring amino acid residues in a beta sheet 2 2.2 Cation interactions with protein backbone oxygen 3 2.3 Quantum molecular dynamics with quantum mechanical treatment of every water molecule 3 3 Methods 4 4 Results 5 4.1 Preparation 5 4.2 Experiment 1302 5 4.3 Experiment 1303 7 5 Discussion 9 5.1 HB networks of water 9 5.2 Subsequent to rupture of a transient beta sheet 9 5.3 Hofmeister effects 10 6 Conclusion 11 7 Future work 12 8 Acknowledgements 13 9 References 14 10 Appendix 1. Backbone hydration in experiment 1302 16 11 Appendix 2. Backbone hydration in experiment 1303 17 1 Abstract It is known that steric blocking by peptide sidechains of hydrogen bonding, HB, between water and peptide groups, PGs, in beta sheets accords with an amino acid intrinsic beta sheet preference [1]. The present observations with Quantum Molecular Dynamics, QMD, simulation with Quantum Mechanical, QM, treatment of every water molecule solvating a beta sheet that would be transient in nature suggest that this steric blocking is not applicable in a hydrophobic region unless a cation is present, so that the amino acid beta sheet preference due to this steric blocking is only effective in the presence of a cation. We observed backbone hydration in a polyalanine and to a lesser extent polyvaline alpha helix without a cation being present, but a cation could increase the strength of these HBs. -
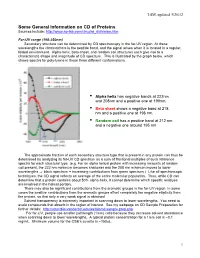
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian
G C A T T A C G G C A T genes Article Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment Edmond Wonkam-Tingang 1, Isabelle Schrauwen 2 , Kevin K. Esoh 1 , Thashi Bharadwaj 2, Liz M. Nouel-Saied 2 , Anushree Acharya 2, Abdul Nasir 3 , Samuel M. Adadey 1,4 , Shaheen Mowla 5 , Suzanne M. Leal 2 and Ambroise Wonkam 1,* 1 Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa; [email protected] (E.W.-T.); [email protected] (K.K.E.); [email protected] (S.M.A.) 2 Center for Statistical Genetics, Sergievsky Center, Taub Institute for Alzheimer’s Disease and the Aging Brain, and the Department of Neurology, Columbia University Medical Centre, New York, NY 10032, USA; [email protected] (I.S.); [email protected] (T.B.); [email protected] (L.M.N.-S.); [email protected] (A.A.); [email protected] (S.M.L.) 3 Synthetic Protein Engineering Lab (SPEL), Department of Molecular Science and Technology, Ajou University, Suwon 443-749, Korea; [email protected] 4 West African Centre for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana, Accra LG 54, Ghana 5 Division of Haematology, Department of Pathology, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa; [email protected] * Correspondence: [email protected]; Tel.: +27-21-4066-307 Received: 28 September 2020; Accepted: 20 October 2020; Published: 23 October 2020 Abstract: DNA samples from five members of a multiplex non-consanguineous Cameroonian family, segregating prelingual and progressive autosomal recessive non-syndromic sensorineural hearing impairment, underwent whole exome sequencing. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Further Confirmation of the Association of SLC12A2 with Non-Syndromic
www.nature.com/jhg ARTICLE OPEN Further confirmation of the association of SLC12A2 with non-syndromic autosomal-dominant hearing impairment Samuel M. Adadey1,2,9, Isabelle Schrauwen3,9, Elvis Twumasi Aboagye1, Thashi Bharadwaj3, Kevin K. Esoh 2, Sulman Basit 4, 3 3 5 2 6 1 Anushree Acharya , Liz M. Nouel-Saied ,✉ Khurram Liaqat , Edmond✉ Wonkam-Tingang , Shaheen Mowla , Gordon A. Awandare , Wasim Ahmad 7, Suzanne M. Leal 3,8 and Ambroise Wonkam 2 © The Author(s) 2021 Congenital hearing impairment (HI) is genetically heterogeneous making its genetic diagnosis challenging. Investigation of novel HI genes and variants will enhance our understanding of the molecular mechanisms and to aid genetic diagnosis. We performed exome sequencing and analysis using DNA samples from affected members of two large families from Ghana and Pakistan, segregating autosomal-dominant (AD) non-syndromic HI (NSHI). Using in silico approaches, we modeled and evaluated the effect of the likely pathogenic variants on protein structure and function. We identified two likely pathogenic variants in SLC12A2, c.2935G>A:p.(E979K) and c.2939A>T:p.(E980V), which segregate with NSHI in a Ghanaian and Pakistani family, respectively. SLC12A2 encodes an ion transporter crucial in the homeostasis of the inner ear endolymph and has recently been reported to be implicated in syndromic and non-syndromic HI. Both variants were mapped to alternatively spliced exon 21 of the SLC12A2 gene. Exon 21 encodes for 17 residues in the cytoplasmatic tail of SLC12A2, is highly conserved between species, and preferentially expressed in cochlear tissues. A review of previous studies and our current data showed that out of ten families with either AD non-syndromic or syndromic HI, eight (80%) had variants within the 17 amino acid residue region of exon 21 (48 bp), suggesting that this alternate domain is critical to the transporter activity in the inner ear.