Evolution of Gene Knockout Strains of E. Coli Reveal Regulatory Architectures Governed by Metabolism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fine–Mapping Identifies NAD–ME1 As a Candidate Underlying a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.07.285429; this version posted September 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Fine–mapping identifies NAD–ME1 as a candidate underlying 2 a major locus controlling temporal variation in primary and 3 specialized metabolism in Arabidopsis 4 5 Marta Francisco1, Daniel J. Kliebenstein2,3, Víctor M. Rodríguez1, Pilar Soengas1, 6 Rosaura Abilleira1, María E. Cartea1 7 1Misión Biológica de Galicia, (MBG-CSIC), P.O. Box 28, 36080, Pontevedra, Spain 8 2Department of Plant Sciences, University of California at Davis, Davis, CA 95616, 9 USA. 10 3DynaMo Center of Excellence, University of Copenhagen, Thorvaldsensvej 40, DK- 11 1871 Frederiksberg C, Denmark. 12 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.09.07.285429; this version posted September 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 13 Summary 14 Plant metabolism is modulated by a complex interplay between internal signals and 15 external cues. A major goal of all quantitative metabolomic studies is to clone the 16 underlying genes to understand the mechanistic basis of this variation. Using fine-scale 17 genetic mapping, in this work we report the identification and initial characterization of 18 NAD-DEPENDENT MALIC ENZYME 1 (NAD-ME1) as the candidate gene underlying 19 the pleiotropic network Met.II.15 QTL controlling variation in plant metabolism and 20 circadian clock outputs in the Bay × Sha Arabidopsis population. -

Profiling Cellular Processes in Adipose Tissue During Weight Loss Using Time Series Gene Expression
G C A T T A C G G C A T genes Article Profiling Cellular Processes in Adipose Tissue during Weight Loss Using Time Series Gene Expression Samar H. K. Tareen 1,* , Michiel E. Adriaens 1,* , Ilja C. W. Arts 1,2, Theo M. de Kok 1,3, Roel G. Vink 4, Nadia J. T. Roumans 4, Marleen A. van Baak 4, Edwin C. M. Mariman 4, Chris T. Evelo 1,5,* and Martina Kutmon 1,5,* 1 Maastricht Centre for Systems Biology (MaCSBio), Maastricht University, 6211ER Maastricht, The Netherlands; [email protected] (I.C.W.A.); [email protected] (T.M.d.K.) 2 Department of Epidemiology, CARIM School for Cardiovascular Diseases, Maastricht University, 6211ER Maastricht, The Netherlands 3 Department of Toxicogenomics, GROW School of Oncology and Developmental Biology, Maastricht University, 6211ER Maastricht, The Netherlands 4 Department of Human Biology, NUTRIM Research School, Maastricht University, 6211ER Maastricht, The Netherlands; [email protected] (R.G.V.); [email protected] (N.J.T.R.); [email protected] (M.A.v.B.); [email protected] (E.C.M.M.) 5 Department of Bioinformatics—BiGCaT, NUTRIM Research School, Maastricht University, 6211ER Maastricht, The Netherlands * Correspondence: [email protected] (S.H.K.T.); [email protected] (M.E.A.); [email protected] (C.T.E.); [email protected] (M.K.) Received: 28 September 2018; Accepted: 22 October 2018; Published: 29 October 2018 Abstract: Obesity is a global epidemic identified as a major risk factor for multiple chronic diseases and, consequently, diet-induced weight loss is used to counter obesity. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Mouse Me1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Me1 Knockout Project (CRISPR/Cas9) Objective: To create a Me1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Me1 gene (NCBI Reference Sequence: NM_001198933 ; Ensembl: ENSMUSG00000032418 ) is located on Mouse chromosome 9. 14 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 14 (Transcript: ENSMUST00000185374). Exon 2~3 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a spontaneous allele exhibit decreased body weight on a medium fat diet, altered cytoplasmic malic enzyme activity, and a male-specific reduction in food intake on a high fat diet. Exon 2 starts from about 1.15% of the coding region. Exon 2~3 covers 17.15% of the coding region. The size of effective KO region: ~2089 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 14 Legends Exon of mouse Me1 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. -

Dual Specificity Phosphatases from Molecular Mechanisms to Biological Function
International Journal of Molecular Sciences Dual Specificity Phosphatases From Molecular Mechanisms to Biological Function Edited by Rafael Pulido and Roland Lang Printed Edition of the Special Issue Published in International Journal of Molecular Sciences www.mdpi.com/journal/ijms Dual Specificity Phosphatases Dual Specificity Phosphatases From Molecular Mechanisms to Biological Function Special Issue Editors Rafael Pulido Roland Lang MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade Special Issue Editors Rafael Pulido Roland Lang Biocruces Health Research Institute University Hospital Erlangen Spain Germany Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal International Journal of Molecular Sciences (ISSN 1422-0067) from 2018 to 2019 (available at: https: //www.mdpi.com/journal/ijms/special issues/DUSPs). For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03921-688-8 (Pbk) ISBN 978-3-03921-689-5 (PDF) c 2019 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. Contents About the Special Issue Editors .................................... -

Dissecting the Genetic Etiology of Lupus at ETS1 Locus
Dissecting the Genetic Etiology of Lupus at ETS1 Locus A dissertation submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Immunobiology of the College of Medicine 2017 by Xiaoming Lu B.S. Sun Yat-sen University, P.R. China June 2011 Dissertation Committee: John B. Harley, MD, PhD Harinder Singh, PhD Leah C. Kottyan, PhD Matthew T. Weirauch, PhD Kasper Hoebe, PhD Lili Ding, PhD i Abstract Systemic lupus erythematosus (SLE) is a complex autoimmune disease with strong evidence for genetics factor involvement. Genome-wide association studies have identified 84 risk loci associated with SLE. However, the specific genotype-dependent (allelic) molecular mechanisms connecting these lupus-genetic risk loci to immunological dysregulation are mostly still unidentified. ~ 90% of these loci contain variants that are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we developed a strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. This strategic approach was then used for investigating SLE association at ETS1 locus. Genetic variants at chromosomal region 11q23.3, near the gene ETS1, have been associated with systemic lupus erythematosus (SLE), or lupus, in independent cohorts of Asian ancestry. -
Supplementary Table 1. Expression
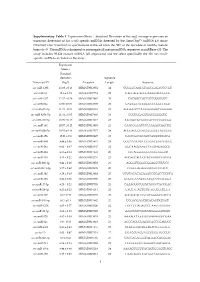
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Mouse Me1 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Me1 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Me1 conditional knockout Mouse model (C57BL/6N) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Me1 gene (NCBI Reference Sequence: NM_001198933 ; Ensembl: ENSMUSG00000032418 ) is located on Mouse chromosome 9. 14 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 14 (Transcript: ENSMUST00000185374). Exon 2 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Me1 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-56G17 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a spontaneous allele exhibit decreased body weight on a medium fat diet, altered cytoplasmic malic enzyme activity, and a male-specific reduction in food intake on a high fat diet. Exon 2 starts from about 1.15% of the coding region. The knockout of Exon 2 will result in frameshift of the gene. The size of intron 1 for 5'-loxP site insertion: 7005 bp, and the size of intron 2 for 3'-loxP site insertion: 1805 bp. The size of effective cKO region: ~634 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 3 14 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Me1 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

ME1 Monoclonal Antibody (M03), Clone 3H5
ME1 monoclonal antibody (M03), clone 3H5 Catalog # : H00004199-M03 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a full length recombinant Western Blot (Cell lysate) Description: ME1. Immunogen: ME1 (AAH25246, 1 a.a. ~ 572 a.a) full-length recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: MEPEAPRRRHTHQRGYLLTRNPHLNKDLAFTLEERQQLNIHGLLPPSFN SQEIQVLRVVKNFEHLNSDFDRYLLLMDLQDRNEKLFYRVLTSDIEKFMPI enlarge VYTPTVGLACQQYSLVFRKPRGLFITIHDRGHIASVLNAWPEDVIKAIVVT Western Blot (Transfected DGERILGLGDLGCNGMGIPVGKLALYTACGGMNPQECLPVILDVGTENE lysate) ELLKDPLYIGLRQRRVRGSEYDDFLDEFMEAVSSKYGMNCLIQFEDFAN VNAFRLLNKYRNQYCTFNDDIQGTASVAVAGLLAALRITKNKLSDQTILFQ GAGEAALGIAHLIVMALEKEGLPKEKAIKKIWLVDSKGLIVKGRASLTQEKE KFAHEHEEMKNLEAIVQEIKPTALIGVAAIGGAFSEQILKDMAAFNERPIIFA LSNPTSKAECSAEQCYKITKGRAIFASGSPFDPVTLPNGQTLYPGQGNN SYVFPGVALGVVACGLRQITDNIFLTTAEVIAQQVSDKHLEEGRLYPPLNT IRDVSLKIAEKIVKDAYQEKTATVYPEPQNKEAFVRSQMYSTDYDQILPDC YSWPEEVQKIQTKVDQ enlarge Host: Mouse Western Blot (Recombinant protein) Reactivity: Human Immunohistochemistry (Formalin/PFA-fixed paraffin- Isotype: IgG1 Kappa embedded sections) Quality Control Antibody Reactive Against Recombinant Protein. Testing: enlarge Sandwich ELISA (Recombinant protein) Western Blot detection against Immunogen (88.66 KDa) . enlarge Storage Buffer: In 1x PBS, pH 7.4 ELISA Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Datasheet: Download Publication Reference 1. Role for malic enzyme, -

Cross-Species Transcriptomic Analysis Elucidates Constitutive Aryl
Sun et al. BMC Genomics 2014, 15:1053 http://www.biomedcentral.com/1471-2164/15/1053 RESEARCH ARTICLE Open Access Cross-species transcriptomic analysis elucidates constitutive aryl hydrocarbon receptor activity Ren X Sun1, Lauren C Chong1, Trent T Simmons1, Kathleen E Houlahan1, Stephenie D Prokopec1, John D Watson1, Ivy D Moffat2, Sanna Lensu3,4, Jere Lindén5, Christine P'ng1, Allan B Okey2, Raimo Pohjanvirta6 and Paul C Boutros1,2,7,8* Abstract Background: Research on the aryl hydrocarbon receptor (AHR) has largely focused on variations in toxic outcomes resulting from its activation by halogenated aromatic hydrocarbons. But the AHR also plays key roles in regulating pathways critical for development, and after decades of research the mechanisms underlying physiological regulation by the AHR remain poorly characterized. Previous studies identified several core genes that respond to xenobiotic AHR ligands across a broad range of species and tissues. However, only limited inferences have been made regarding its role in regulating constitutive gene activity, i.e. in the absence of exogenous ligands. To address this, we profiled transcriptomic variations between AHR-active and AHR-less-active animals in the absence of an exogenous agonist across five tissues, three of which came from rats (hypothalamus, white adipose and liver) and two of which came from mice (kidney and liver). Because AHR status alone has been shown sufficient to alter transcriptomic responses, we reason that by contrasting profiles amongst AHR-variant animals, we may elucidate effects of the AHR on constitutive mRNA abundances. Results: We found significantly more overlap in constitutive mRNA abundances amongst tissues within the same species than from tissues between species and identified 13 genes (Agt, Car3, Creg1, Ctsc, E2f6, Enpp1, Gatm, Gstm4, Kcnj8, Me1, Pdk1, Slc35a3,andSqrdl) that are affected by AHR-status in four of five tissues. -

Discovery of Gene Sources for Economic Traits in Hanwoo by Whole-Genome Resequencing
1353 Open Access Asian Australas. J. Anim. Sci. Vol. 29, No. 9 : 1353-1362 September 2016 http://dx.doi.org/10.5713/ajas.15.0760 www.ajas.info pISSN 1011-2367 eISSN 1976-5517 Discovery of Gene Sources for Economic Traits in Hanwoo by Whole-genome Resequencing Younhee Shin, Ho-jin Jung, Myunghee Jung, Seungil Yoo, Sathiyamoorthy Subramaniyam, Kesavan Markkandan, Jun-Mo Kang1, Rajani Rai1, Junhyung Park*, and Jong-Joo Kim1,* Codes Division, Insilicogen Inc., Yongin 446-908, Korea ABSTRACT: Hanwoo, a Korean native cattle (Bos taurus coreana), has great economic value due to high meat quality. Also, the breed has genetic variations that are associated with production traits such as health, disease resistance, reproduction, growth as well as carcass quality. In this study, next generation sequencing technologies and the availability of an appropriate reference genome were applied to discover a large amount of single nucleotide polymorphisms (SNPs) in ten Hanwoo bulls. Analysis of whole-genome resequencing generated a total of 26.5 Gb data, of which 594,716,859 and 592,990,750 reads covered 98.73% and 93.79% of the bovine reference genomes of UMD 3.1 and Btau 4.6.1, respectively. In total, 2,473,884 and 2,402,997 putative SNPs were discovered, of which 1,095,922 (44.3%) and 982,674 (40.9%) novel SNPs were discovered against UMD3.1 and Btau 4.6.1, respectively. Among the SNPs, the 46,301 (UMD 3.1) and 28,613 SNPs (Btau 4.6.1) that were identified as Hanwoo-specific SNPs were included in the functional genes that may be involved in the mechanisms of milk production, tenderness, juiciness, marbling of Hanwoo beef and yellow hair.