Mining Patterns in Genomic and Clinical Cancer Data to Characterize Novel Driver Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Unc93b Antibody (Pab)
21.10.2014Unc93b antibody (pAb) Rabbit Anti -Human/Mouse/Rat Unc93b Instruction Manual Catalog Number PK-AB718-4553 Synonyms Unc93b Antibody: Unc93b1, homolog of C. elegans Unc93 Description The endoplasmic reticulum (ER) protein Unc93b, a human homolog of the C. elegans Unc93 gene, was initially identified by a forward genetic screen using N-ethyl-N-nitrosourea where a histidine- to-arginine substitution in Unc93b caused defects in Toll-like receptor (TLR) 3, 7 and 9 signaling. Unlike Unc93a, another homolog of the C. elegans Unc93 gene whose function is unknown, Unc93b specifically interacts with TLR3, 7 and 9; the histidine-to-arginine point mutation used to identify Unc93b abolishes this interaction. Mice carrying this point mutation are highly susceptible to infection with a number of viruses, indicating that Unc93b plays an important role in innate immunity. Multiple isoforms of Unc93a are known to exist. This antibody will not cross-react with Unc93a. Quantity 100 µg Source / Host Rabbit Immunogen Unc93b antibody was raised in rabbits against a 19 amino acid peptide from near the amino terminus of human Unc93b. Purification Method Affinity chromatography purified via peptide column. Clone / IgG Subtype Polyclonal antibody Species Reactivity Human, Mouse, Rat Specificity Multiple isoforms of Unc93a are known to exist. This antibody will not cross-react with Unc93a. Formulation Antibody is supplied in PBS containing 0.02% sodium azide. Reconstitution During shipment, small volumes of antibody will occasionally become entrapped in the seal of the product vial. For products with volumes of 200 μl or less, we recommend gently tapping the vial on a hard surface or briefly centrifuging the vial in a tabletop centrifuge to dislodge any liquid in the container’s cap. -

Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer
Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer By Josiah Ewing Hutton, III. Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biochemistry December 2016 Nashville, Tennessee Approved: Daniel C. Liebler, Ph.D. Robert J. Coffey, M.D. Bruce D. Carter, Ph.D. Nicholas J. Reiter, Ph.D. Jamey D. Young, Ph.D. Charles R. Sanders, Ph.D. Acknowledgements I would like to acknowledge my mentor, Dr. Daniel Liebler, for his constant guidance throughout my graduate career. Dan allowed me the freedom to develop as a scientist, and tactfully provided the tutelage and discussion I needed at exactly the right time and not a moment earlier. I will be grateful for his mentorship, insight, and his wit for the rest of my career. I would also like to acknowledge all current and former laboratory members for not only providing insight and guidance with my dissertation, but also for making every single day in lab enjoyable. I wish to acknowledge my committee members, Dr. Bruce Carter, Dr. Robert Coffey, Dr. Nicholas Reiter, Dr. Charles Sanders, and Dr. Jamey Young. Our meetings throughout the years have helped to guide my research to the work that it is today. I am grateful to my friends, who have always been there for me and would quietly, and sometimes not so quietly, listen to me rehearse my dissertation while we were climbing, so long as I would then push them to climb harder. Lastly, I am grateful to my family, who have always supported me and driven me to work harder. -

Analysis of the Human Serum Proteome
Cedarville University DigitalCommons@Cedarville Pharmaceutical Sciences Faculty Publications Department of Pharmaceutical Sciences 6-2004 Analysis of the Human Serum Proteome King C. Chan David A. Lucas Denise Hise Carl F. Schaefer Zhen Xiao See next page for additional authors Follow this and additional works at: https://digitalcommons.cedarville.edu/ pharmaceutical_sciences_publications Part of the Pharmacy and Pharmaceutical Sciences Commons This Article is brought to you for free and open access by DigitalCommons@Cedarville, a service of the Centennial Library. It has been accepted for inclusion in Pharmaceutical Sciences Faculty Publications by an authorized administrator of DigitalCommons@Cedarville. For more information, please contact [email protected]. Authors King C. Chan, David A. Lucas, Denise Hise, Carl F. Schaefer, Zhen Xiao, George M. Janini, Kenneth H. Buetow, Haleem J. Issaq, Timothy D. Veenstra, and Thomas P. Conrads Clinical Proteomics Journal Copyright ©Humana Press Inc. All rights of any nature whatsoever are reserved. ISSN 1542-6416/04/01:101–225/$25.00 Serum/Plasma Proteome Analysis of the Human Serum Proteome King C. Chan,1,† David A. Lucas,1,† Denise Hise,2 Carl F. Schaefer,2 Zhen Xiao,1 George M. Janini,1 Kenneth H. Buetow,2 Haleem J. Issaq,1 Timothy D.Veenstra,1 and Thomas P. Conrads1,* 1Laboratory of Proteomics and Analytical Technologies, National Cancer Institute at Frederick, SAIC-Frederick, Inc, PO Box B, Frederick, MD 21702 2Center for Bioinformatics, National Cancer Institute, Bethesda, MD 20892 †These authors contributed equally to this work. each of which was analyzed by microcapillary Abstract reversed-phase liquid chromatography coupled Changes in serum proteins that signal online with MS/MS analysis. -

Interoperability in Toxicology: Connecting Chemical, Biological, and Complex Disease Data
INTEROPERABILITY IN TOXICOLOGY: CONNECTING CHEMICAL, BIOLOGICAL, AND COMPLEX DISEASE DATA Sean Mackey Watford A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Gillings School of Global Public Health (Environmental Sciences and Engineering). Chapel Hill 2019 Approved by: Rebecca Fry Matt Martin Avram Gold David Reif Ivan Rusyn © 2019 Sean Mackey Watford ALL RIGHTS RESERVED ii ABSTRACT Sean Mackey Watford: Interoperability in Toxicology: Connecting Chemical, Biological, and Complex Disease Data (Under the direction of Rebecca Fry) The current regulatory framework in toXicology is expanding beyond traditional animal toXicity testing to include new approach methodologies (NAMs) like computational models built using rapidly generated dose-response information like US Environmental Protection Agency’s ToXicity Forecaster (ToXCast) and the interagency collaborative ToX21 initiative. These programs have provided new opportunities for research but also introduced challenges in application of this information to current regulatory needs. One such challenge is linking in vitro chemical bioactivity to adverse outcomes like cancer or other complex diseases. To utilize NAMs in prediction of compleX disease, information from traditional and new sources must be interoperable for easy integration. The work presented here describes the development of a bioinformatic tool, a database of traditional toXicity information with improved interoperability, and efforts to use these new tools together to inform prediction of cancer and complex disease. First, a bioinformatic tool was developed to provide a ranked list of Medical Subject Heading (MeSH) to gene associations based on literature support, enabling connection of compleX diseases to genes potentially involved. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
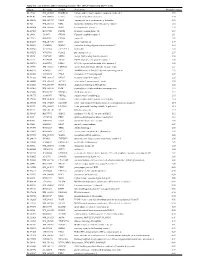
Supplementary Table 1
Table S1. List of Genes Differentially Expressed in YB-1 siRNA-Transfected MCF-7 Cells Unigene Accession Symbol Description Mean fold change Hs.17466 NM_004585 RARRES3 retinoic acid receptor responder (tazarotene induced) 3 3.57 Hs.64746 NM_004669 CLIC3 chloride intracellular channel 3 3.33 Hs.516155 NM_001747 CAPG capping protein (actin filament), gelsolin-like 2.88 Hs.926 NM_002463 MX2 myxovirus (influenza virus) resistance 2 (mouse) 2.72 Hs.643494 NM_005410 SEPP1 selenoprotein P, plasma, 1 2.70 Hs.267038 BC017500 POF1B premature ovarian failure 1B 2.67 Hs.20961 U63917 GPR30 G protein- coupled receptor 30 2532.53 Hs.199877 BE645967 CPNE4 copine IV 2.49 Hs.632177 NM_017458 MVP major vault protein 2.48 Hs.528836 AA005023 NOD27 nucleotide-binding oligomerization domains 27 2.43 Hs.360940 AL589866 dJ222E13.1 kraken-like 2.40 Hs.515575 AI743780 PLAC2 placenta-specific 2 2.37 Hs.25674 AI827820 MBD2 methyl-CpG binding domain protein 2 2.36 Hs.12229 AA149594 TIEG2 TGFB inducible early growth response 2 2.33 Hs.482730 AA053711 EDIL3 EGF-like repeats and discoidin I-like domains 3 2.32 Hs. 370771 NM_000389 CDKN1A cyclin- depen dent kinase in hibitor 1A (p 21, Cip 1) 2282.28 Hs.469175 AI341537 JFC1 NADPH oxidase-related, C2 domain-containing protein 2.28 Hs.514821 AF043341 CCL5 chemokine (C-C motif) ligand 5 2.27 Hs.591292 NM_023915 GPR87 G protein-coupled receptor 87 2.26 Hs.500483 NM_001613 ACTA2 actin, alpha 2, smooth muscle, aorta 2.24 Hs.632824 NM_006729 DIAPH2 diaphanous homolog 2 (Drosophila) 2.22 Hs.369430 NM_000919 PAM peptidylglycine -

Laboratory Genetic Testing in Clinical Practice
BioMed Research International Laboratory Genetic Testing in Clinical Practice Guest Editors: Ozgur Cogulu, Yasemin Alanay, and Gokce A. Toruner Laboratory Genetic Testing in Clinical Practice BioMed Research International Laboratory Genetic Testing in Clinical Practice Guest Editors: Ozgur Cogulu, Yasemin Alanay, and Gokce A. Toruner Copyright © 2013 Hindawi Publishing Corporation. All rights reserved. This is a special issue published in “BioMed Research International.” All articles are open access articles distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Contents Laboratory Genetic Testing in Clinical Practice,OzgurCogulu,YaseminAlanay,andGokceA.Toruner Volume 2013, Article ID 532897, 1 page Identification and Characterization of DM1 Patients by a NewDiagnostic Certified Assay: Neuromuscular and Cardiac Assessments, Rea Valaperta, Valeria Sansone, Fortunata Lombardi, Chiara Verdelli, Alessio Colombo, Massimiliano Valisi, Elisa Brigonzi, Elena Costa, and Giovanni Meola Volume 2013, Article ID 958510, 6 pages Ultradeep Pyrosequencing of Hepatitis C Virus Hypervariable Region 1 in Quasispecies Analysis, Kamila Caraballo Cortes,´ Osvaldo Zagordi, Tomasz Laskus, Rafal Ploski, Iwona Bukowska-O´sko, Agnieszka Pawelczyk, Hanna Berak, and Marek Radkowski Volume 2013, Article ID 626083, 10 pages Feasibility of a Microarray-Based Point-of-Care CYP2C19 Genotyping Test for Predicting Clopidogrel On-Treatment Platelet -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

The Role of Cyclin B3 in Mammalian Meiosis
THE ROLE OF CYCLIN B3 IN MAMMALIAN MEIOSIS by Mehmet Erman Karasu A Dissertation Presented to the Faculty of the Louis V. Gerstner Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy New York, NY November, 2018 Scott Keeney, PhD Date Dissertation Mentor Copyright © Mehmet Erman Karasu 2018 DEDICATION I would like to dedicate this thesis to my parents, Mukaddes and Mustafa Karasu. I have been so lucky to have their support and unconditional love in this life. ii ABSTRACT Cyclins and cyclin dependent kinases (CDKs) lie at the center of the regulation of the cell cycle. Cyclins as regulatory partners of CDKs control the switch-like cell cycle transitions that orchestrate orderly duplication and segregation of genomes. Similar to somatic cell division, temporal regulation of cyclin-CDK activity is also important in meiosis, which is the specialized cell division that generates gametes for sexual production by halving the genome. Meiosis does so by carrying out one round of DNA replication followed by two successive divisions without another intervening phase of DNA replication. In budding yeast, cyclin-CDK activity has been shown to have a crucial role in meiotic events such as formation of meiotic double-strand breaks that initiate homologous recombination. Mammalian cells express numerous cyclins and CDKs, but how these proteins control meiosis remains poorly understood. Cyclin B3 was previously identified as germ cell specific, and its restricted expression pattern at the beginning of meiosis made it an interesting candidate to regulate meiotic events. -

Kinase Profiling Book
Custom and Pre-Selected Kinase Prof iling to f it your Budget and Needs! As of July 1, 2021 19.8653 mm 128 196 12 Tyrosine Serine/Threonine Lipid Kinases Kinases Kinases Carna Biosciences, Inc. 2007 Carna Biosciences, Inc. Profiling Assays available from Carna Biosciences, Inc. As of July 1, 2021 Page Kinase Name Assay Platform Page Kinase Name Assay Platform 4 ABL(ABL1) MSA 21 EGFR[T790M/C797S/L858R] MSA 4 ABL(ABL1)[E255K] MSA 21 EGFR[T790M/L858R] MSA 4 ABL(ABL1)[T315I] MSA 21 EPHA1 MSA 4 ACK(TNK2) MSA 21 EPHA2 MSA 4 AKT1 MSA 21 EPHA3 MSA 5 AKT2 MSA 22 EPHA4 MSA 5 AKT3 MSA 22 EPHA5 MSA 5 ALK MSA 22 EPHA6 MSA 5 ALK[C1156Y] MSA 22 EPHA7 MSA 5 ALK[F1174L] MSA 22 EPHA8 MSA 6 ALK[G1202R] MSA 23 EPHB1 MSA 6 ALK[G1269A] MSA 23 EPHB2 MSA 6 ALK[L1196M] MSA 23 EPHB3 MSA 6 ALK[R1275Q] MSA 23 EPHB4 MSA 6 ALK[T1151_L1152insT] MSA 23 Erk1(MAPK3) MSA 7 EML4-ALK MSA 24 Erk2(MAPK1) MSA 7 NPM1-ALK MSA 24 Erk5(MAPK7) MSA 7 AMPKα1/β1/γ1(PRKAA1/B1/G1) MSA 24 FAK(PTK2) MSA 7 AMPKα2/β1/γ1(PRKAA2/B1/G1) MSA 24 FER MSA 7 ARG(ABL2) MSA 24 FES MSA 8 AurA(AURKA) MSA 25 FGFR1 MSA 8 AurA(AURKA)/TPX2 MSA 25 FGFR1[V561M] MSA 8 AurB(AURKB)/INCENP MSA 25 FGFR2 MSA 8 AurC(AURKC) MSA 25 FGFR2[V564I] MSA 8 AXL MSA 25 FGFR3 MSA 9 BLK MSA 26 FGFR3[K650E] MSA 9 BMX MSA 26 FGFR3[K650M] MSA 9 BRK(PTK6) MSA 26 FGFR3[V555L] MSA 9 BRSK1 MSA 26 FGFR3[V555M] MSA 9 BRSK2 MSA 26 FGFR4 MSA 10 BTK MSA 27 FGFR4[N535K] MSA 10 BTK[C481S] MSA 27 FGFR4[V550E] MSA 10 BUB1/BUB3 MSA 27 FGFR4[V550L] MSA 10 CaMK1α(CAMK1) MSA 27 FGR MSA 10 CaMK1δ(CAMK1D) MSA 27 FLT1 MSA 11 CaMK2α(CAMK2A) MSA 28