Chapter 1 Synthesis of (±)-Baclofen and Analogs (Homobaclofen, PCPGABA, Phaclofen, Saclofen)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

(12) Patent Application Publication (10) Pub. No.: US 2009/0156618 A1 Tollefson (43) Pub
US 20090156618A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0156618 A1 Tollefson (43) Pub. Date: Jun. 18, 2009 (54) 1-(1-(2-ETHOXYETHYL)-3-ETHYL-7- Related U.S. Application Data (4-METHYLPYRIDIN-2-YLAMINO) - 1H-PYRAZOLO 4.3-D PYRIMIDIN-5-YL) (60) Provisional application No. 60/735,320, filed on Nov. PPERDINE-4-CARBOXYLIC ACID AND 10, 2005. SALTS THEREOF Publication Classification (51) Int. Cl. (75) Inventor: Michael B. Tollefson, Dardenne A 6LX 3/59 (2006.01) Prairie, MO (US) C07D 487/04 (2006.01) A6IP 9/12 (2006.01) A6IP 25/00 (2006.01) Correspondence Address: A6IP 9/00 (2006.01) PFZER INC. PATENT DEPARTMENT, Bld 114 M/S 114, (52) U.S. Cl. ...................................... 514/262.1; 54.4/262 EASTERN PONT ROAD (57) ABSTRACT GROTON, CT 06340 (US) The present invention comprises 1-(1-(2-ethoxyethyl)-3- ethyl-7-(4-methylpyridin-2-ylamino)-1H-pyrazolo 4.3-d (73) Assignee: Pfizer Inc pyrimidin-5-yl)piperidine-4-carboxylic acid and its salts. The invention further comprises pharmaceutical composi tions, methods of treatment, and synthetic methods relating to (21) Appl. No.: 111558,306 1-(1-(2-ethoxyethyl)-3-ethyl-7-(4-methylpyridin-2- ylamino)-1H-pyrazolo 4.3-dipyrimidin-5-yl)piperidine-4- (22) Filed: Nov. 9, 2006 carboxylic acid and its salts. Patent Application Publication Jun. 18, 2009 Sheet 2 of 2 US 2009/0156618 A1 "SOIHZ US 2009/0156618 A1 Jun. 18, 2009 1-(1-(2-ETHOXYETHYL)-3-ETHYL-7- 1H-pyrazolo 4,3-dipyrimidin-5-yl)piperidine-4-carboxylic (4-METHYLPYRIDIN-2-YLAMINO) - acid and its pharmaceutically acceptable salts. -

Determining the Role of Gabaergic Signaling in The
DETERMINING THE ROLE OF GABAERGIC SIGNALING IN THE CRANIOFACIAL DEVELOPMENT OF LARVAL ZEBRAFISH by LINDSEY L. BEEBE (Under the Direction of James D. Lauderdale) ABSTRACT Although best known as an inhibitory neurotransmitter, intriguing evidence has implicated GABA as a key signaling molecule in craniofacial development in mammals. Glutamate is converted to GABA by an enzyme called glutamic acid decarboxylase (GAD), which exists in two isoforms, GAD67 and GAD65. The GAD1 and GAD2 genes encode these isoforms, respectively. A decrease in GAD activity in the human brain is often associated with epilepsy, schizophrenia and related neurological disorders. In mice and humans, mutations in gad1, but not gad2, result in defects in palate development, and mutations in the Gabrb3 gene, which encodes the β3 subunit of the GABAA receptor, exhibit a comparable phenotype to gad1 mutations. These results suggest that GABA signaling, through the GABAA receptor, can play an important and conserved role in craniofacial development. However, the mechanism of this process is not known and cannot be easily investigated in a mammalian system. In this work, translation-blocking morpholinos against the GAD genes were used to alter expression within the larval zebrafish. While gad2 morphants looked phenotypically normal, gad1 morphant animals exhibited altered cranial structures at 1 and 7 dpf. Yet, both gad1 and gad2 morphants exhibited spontaneous seizure-like neural activity. Through the use of photoactivatable caged-morpholinos, the craniofacial deformities could be bypassed when photolysis was carried out at 24 hpf. Electrophysiological recordings showed that while dark-raised CyHQ-gad1 morphant animals looked phenotypically comparable to wild-type animals, they exhibited abnormal, seizure-like neural activity. -
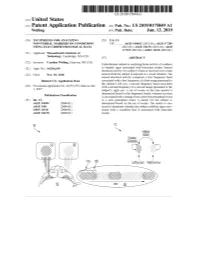
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Campro Catalog Stable Isotope
Introduction & Welcome Dear Valued Customer, We are pleased to present to you our Stable Isotopes Catalog which contains more than three thousand (3000) high quality labeled compounds. You will find new additions that are beneficial for your research. Campro Scientific is proud to work together with Isotec, Inc. for the distribution and marketing of their stable isotopes. We have been working with Isotec for more than twenty years and know that their products meet the highest standard. Campro Scientific was founded in 1981 and we provide services to some of the most prestigious universities, research institutes and laboratories throughout Europe. We are a research-oriented company specialized in supporting the requirements of the scientific community. We are the exclusive distributor of some of the world’s leading producers of research chemicals, radioisotopes, stable isotopes and environmental standards. We understand the requirements of our customers, and work every day to fulfill them. In working with us you are guaranteed to receive: - Excellent customer service - High quality products - Dependable service - Efficient distribution The highly educated staff at Campro’s headquarters and sales office is ready to assist you with your questions and product requirements. Feel free to call us at any time. Sincerely, Dr. Ahmad Rajabi General Manager 180/280 = unlabeled 185/285 = 15N labeled 181/281 = double labeled (13C+15N, 13C+D, 15N+18O etc.) 186/286 = 12C labeled 182/282 = d labeled 187/287 = 17O labeled 183/283 = 13C labeleld 188/288 = 18O labeled 184/284 = 16O labeled, 14N labeled 189/289 = Noble Gases Table of Contents Ordering Information.................................................................................................. page 4 - 5 Packaging Information .............................................................................................. -

(12) Patent Application Publication (10) Pub. No.: US 2007/0254920 A1 Delong Et Al
US 20070254920A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2007/0254920 A1 deLong et al. (43) Pub. Date: Nov. 1, 2007 (54) PRODRUG DERIVATIVES OF ACIDS USING (52) U.S. Cl. ......................... 514/319; 514/323: 514/573; ALCOHOLS WITH HOMOTOPIC HYDROXY 514/415: 514/443; 514/469; GROUPS AND METHODS FOR THEIR 546/196; 546/201: 546/206; PREPARATION AND USE 548/495; 549/49; 549/462 (75) Inventors: Mitchell A. deLong, Raleigh, NC (US); (57) ABSTRACT Jill M. McFadden, Chapel Hill, NC This invention relates to novel homotopic prodrugs and (US); Susan M. Royalty, Cary, NC medicaments and methods for their preparation, testing and (US); Eric J. Toone, Durham, NC use. In one embodiment, the homotopic prodrug has the (US); Jeffrey D. Yingling, Apex, NC general formula (US) Correspondence Address: O MICHAEL BEST & FRIEDRCH LLP 100 E WISCONSNAVENUE Rb, Suite 3300 --- MILWAUKEE, WI 53202 (US) wherein (73) Assignee: Aerie Pharmaceuticals, Inc., Research Triangle Park, NC (21) Appl. No.: 11/412,207 (22) Filed: Apr. 26, 2006 -- Publication Classification is a biologically-active moiety comprising a carboxylic acid functional group, and R is a homotopically-symmetrical (51) Int. Cl. alcohol bonded to the biologically-active moiety through the A6 IK 3/558 (2006.01) carboxylic acid functional group to form an ester linkage, as CO7D 409/02 (2006.01) well as optical isomers, enantiomers, pharmaceutically CO7D 405/02 (2006.01) acceptable salts, biohydrolyzable amides, esters, and imides CO7D 403/02 (2006.01) thereof and combinations thereof. US 2007/0254920 A1 Nov. 1, 2007 PRODRUG DERVATIVES OF ACDS USING WO 99/12896, and WO 99/12898.) However, such modifi ALCOHOLS WITH HOMOTOPIC HYDROXY cations have either resulted in only modest increases in GROUPS AND METHODS FOR THEIR half-life or resulted in compounds with diminished potency. -

(12) United States Patent (10) Patent No.: US 7,304,086 B2 Schilling Et Al
USOO7304086B2 (12) United States Patent (10) Patent No.: US 7,304,086 B2 Schilling et al. (45) Date of Patent: *Dec. 4, 2007 (54) INHIBITORS OF GLUTAMINYL CYCLASE WO WOOO,53596 9, 2000 WO WO O1/34594 A1 5, 2001 (75) Inventors: Stephan Schilling, Halle/Saale (DE); WO WO O2, 13821 2, 2002 WO WO O2, 16318 A1 2, 2002 Mirko Buchholz, Halle/Saale (DE): WO WO O2O16318 A1 * 2/2002 Andre Johannes Niestroj, Sennewitz WO WO O2/O66459 A1 8, 2002 (DE); Ulrich Heiser, Halle/Saale (DE): WO WO O2/O92103 A1 11 2002 Hans-Ulrich Demuth, Halle/Saale (DE) WO WO O2O94813 A1 * 11/2002 WO WO 03/04.0174 A2 5, 2003 (73) Assignee: Probiodrug AG, Halle/Saale (DE) WO WO O3,O70732 8, 2003 WO WO O3068738 8, 2003 (*) Notice: Subject to any disclaimer, the term of this WO WO O30954.21 A1 * 11/2003 patent is extended or adjusted under 35 WO WO 2004O26815 A2 * 4, 2004 U.S.C. 154(b) by 20 days. WO WO 2004/089366 10, 2004 WO WO 2004/098591 A2 11 2004 This patent is Subject to a terminal dis WO WO 2004/098.625 A2 11 2004 claimer. OTHER PUBLICATIONS (21) Appl. No.: 11/051,760 Haley et al. Giorn. Ital. Chemioterap. (1962) 6-9 (No. 3), 213-24. Abstract from CAS attached. (22) Filed: Feb. 4, 2005 Hough et al. Pharmacology, Biochemistry and Behavior 1999, 65(1), 61-66. * Abstract from CAS attached.* (65) Prior Publication Data Morissette et al. Advanced Drug Delivery Reviews 2004, 56. US 2005/0215573 A1 Sep. -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Cell Surface Mobility of GABAB Receptors Saad Bin
Cell surface mobility of GABAB receptors Saad Bin Hannan September 2011 A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy of the University College London Department of Neuroscience, Physiology, and Pharmacology University College London Gower Street London WC1E 6BT UK Declaration ii ‘I, Saad Hannan confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis.' ____________________ Saad Hannan September 2011 To Ammu, Abbu, Polu Abstract ivi Abstract Type-B γ-aminobutyric acid receptors (GABABRs) are important for mediating slow inhibition in the central nervous system and the kinetics of their internalisation and lateral mobility will be a major determinant of their signalling efficacy. Functional GABABRs require R1 and R2 subunit co-assembly, but how heterodimerisation affects the trafficking kinetics of GABABRs is unknown. Here, an α- bungarotoxin binding site (BBS) was inserted into the N-terminus of R2 to monitor receptor mobility in live cells. GABABRs are internalised via clathrin- and dynamin- dependent pathways and recruited to endosomes. By mutating the BBS, a new technique was developed to differentially track R1a and R2 simultaneously, revealing the subunits internalise as heteromers and that R2 dominantly-affects constitutive internalisation of GABABRs. Notably, the internalisation profile of R1aR2 heteromers, but not R1a homomers devoid of their ER retention motif (R1ASA), is similar to R2 homomers in heterologous systems. The internalisation of R1aASA was slowed to that of R2 by mutating a di-leucine motif in the R1 C-terminus, indicating a new role for heterodimerisation, whereby R2 subunits slow the internalization of surface GABABRs. -

Organic Seminar Abstracts
L I B RA FLY OF THE U N IVERSITY OF 1LLI NOIS Q.54-1 Ii£s 1964/65 ,t.l P Return this book on or before the Latest Date stamped below. Theft, mutilation, and underlining of books are reasons for disciplinary action and may result in dismissal from the University. University of Illinois Library OCfcfcsrm L161— O-1096 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/organicsemi1964651univ s ORGANIC SEMINAR ABSTRACTS 196^-65 Semester I Department of Chemistry and Chemical Engineering University of Illinois ' / SEMINAR TOPICS I Semester I96U-I965 f"/ ( Orientation in Sodium and Potassium Metalations of Aromatic Compounds Earl G. Alley 1 Structure of Cyclopropane Virgil Weiss 9 Vinylidenes and Vinylidenecarbenes Joseph C. Catlin 17 Diazene Intermediates James A. Bonham 25 Perfluoroalkyl and Polyfluoroalkyl Carbanions J. David Angerer 3^ Free Radical Additions to Allenes Raymond Feldt 43 The Structure and Biosynthesis of Quassin Richard A. Larson 52 The Decomposition of Perester Compounds Thomas Sharpe 61 The Reaction of Di-t -Butyl Peroxide with Simple Alkyl, Benzyl and Cyclic Ethers R. L. Keener 70 Rearrangements and Solvolysis in Some Allylic Systems Jack Timberlake 79 Longifolene QQ Michael A. Lintner 00 Hydrogenation ! Homogeneous Catalytic Robert Y. Ning 9° c c Total Synthesis of ( -) -Emetime R. Lambert 1* Paracyclophanes Ping C. Huang 112 Mechanism of the Thermal Rearrangement of Cyclopropane George Su 121 Some Recent Studies of the Photochemistry of Cross -Conjugated Cyclohexadienones Elizabeth McLeister 130 The Hammett Acidity Function R. P. Quirk 139 Homoaromaticity Roger A. -

A Review of Glutamate Receptors I: Current Understanding of Their Biology
J Toxicol Pathol 2008; 21: 25–51 Review A Review of Glutamate Receptors I: Current Understanding of Their Biology Colin G. Rousseaux1 1Department of Pathology and Laboratory Medicine, Faculty of Medicine, University of Ottawa, Ottawa, Ontario, Canada Abstract: Seventy years ago it was discovered that glutamate is abundant in the brain and that it plays a central role in brain metabolism. However, it took the scientific community a long time to realize that glutamate also acts as a neurotransmitter. Glutamate is an amino acid and brain tissue contains as much as 5 – 15 mM glutamate per kg depending on the region, which is more than of any other amino acid. The main motivation for the ongoing research on glutamate is due to the role of glutamate in the signal transduction in the nervous systems of apparently all complex living organisms, including man. Glutamate is considered to be the major mediator of excitatory signals in the mammalian central nervous system and is involved in most aspects of normal brain function including cognition, memory and learning. In this review, the basic biology of the excitatory amino acids glutamate, glutamate receptors, GABA, and glycine will first be explored. In the second part of this review, the known pathophysiology and pathology will be described. (J Toxicol Pathol 2008; 21: 25–51) Key words: glutamate, glycine, GABA, glutamate receptors, ionotropic, metabotropic, NMDA, AMPA, review Introduction and Overview glycine), peptides (vasopressin, somatostatin, neurotensin, etc.), and monoamines (norepinephrine, dopamine and In the first decades of the 20th century, research into the serotonin) plus acetylcholine. chemical mediation of the “autonomous” (autonomic) Glutamatergic synaptic transmission in the mammalian nervous system (ANS) was an area that received much central nervous system (CNS) was slowly established over a research activity.