Target Identification Reveals Lanosterol Synthase As a Vulnerability in Glioma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

Anaerobic Bacteria Need Their Vitamin B12 to Digest Estrogen COMMENTARY Montserrat El´Ias-Arnanza,1
COMMENTARY Anaerobic bacteria need their vitamin B12 to digest estrogen COMMENTARY Montserrat El´ıas-Arnanza,1 Often described as nature’s most beautiful cofactor (1), vitamin B12 (cobalamin) is a complex and fascinating organometallic molecule that, although made only by some prokaryotes, has key functional roles in microbes, animals, and humans (2). Its two major biological forms, methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl), have a central cobalt atom in a corrin ring that is coordinated via a cobalt–carbon bond to an upper axial methyl or 5′-deoxyadenosyl group, re- spectively; in both forms the lower axial ligand is the 5,6-dimethylbenzimidazole (DMB) base of a nu- cleotide tail attached to the corrin ring (2–5). Finely controlled enzymatic or photolytic cleavage of the upper axial cobalt–carbon bond underlies the use of B12 as an enzyme cofactor or, in a new twist, as the light-sensing molecule in a photoreceptor pro- tein (2–5). As an enzyme cofactor, MeCbl is used for methyl transfer reactions by methyltransferases and Fig. 1. Proposed B12-dependent methylation of estrogen to androgen in AdoCbl for radical-based transformations by mutases, anaerobic denitrifying bacteria. The putative catalytic and B12-binding units are EmtA and EmtB, respectively, which are encoded by the estrogen–up-regulated dehydratases, deaminases, and ribonucleotide reduc- emtABCD operon. As in well-characterized B -dependent methyltransferases, the – 12 tases (2 4). In a broad range of organisms and bio- cobalt in B12 is expected to cycle between the +3and+1oxidationstates.The logical processes, B12-dependent methyltransferases latter is a potent nucleophile that suffers occasional oxidative inactivation to the catalyze the transfer of a methyl group from a donor +2 state and requires reductive activation to reenter the catalytic cycle (dotted to a final acceptor, utilizing MeCbl or its methylcobamide arrows). -

Aquatic Toxicology 176 (2016) 116–127
Aquatic Toxicology 176 (2016) 116–127 Contents lists available at ScienceDirect Aquatic Toxicology j ournal homepage: www.elsevier.com/locate/aquatox Linking the response of endocrine regulated genes to adverse effects on sex differentiation improves comprehension of aromatase inhibition in a Fish Sexual Development Test a,∗ b b b Elke Muth-Köhne , Kathi Westphal-Settele , Jasmin Brückner , Sabine Konradi , c a a,1 d,1 Viktoria Schiller , Christoph Schäfers , Matthias Teigeler , Martina Fenske a Fraunhofer IME, Department of Ecotoxicology, Auf dem Aberg 1, 57392 Schmallenberg, Germany b German Environment Agency (UBA), Woerlitzer Platz 1, 06844 Dessau, Germany c Fraunhofer IME, Attract Group UNIFISH, Forckenbeckstraße 6, 52074 Aachen, Germany d Fraunhofer IME, Project Group Translational Medicine and Pharmacology TMP, Forckenbeckstraße 6, 52074 Aachen, Germany a r a t i c l e i n f o b s t r a c t Article history: The Fish Sexual Development Test (FSDT) is a non-reproductive test to assess adverse effects of endocrine Received 23 December 2015 disrupting chemicals. With the present study it was intended to evaluate whether gene expression end- Received in revised form 13 April 2016 points would serve as predictive markers of endocrine disruption in a FSDT. For proof-of-concept, a FSDT Accepted 19 April 2016 according to the OECD TG 234 was conducted with the non-steroidal aromatase inhibitor fadrozole (test Available online 20 April 2016 concentrations: 10 g/L, 32 g/L, 100 g/L) using zebrafish (Danio rerio). Gene expression analyses using quantitative RT-PCR were included at 48 h, 96 h, 28 days and 63 days post fertilization (hpf, dpf). -

Friedelin Synthase from Maytenus Ilicifolia
www.nature.com/scientificreports OPEN Friedelin Synthase from Maytenus ilicifolia: Leucine 482 Plays an Essential Role in the Production of Received: 09 May 2016 Accepted: 20 October 2016 the Most Rearranged Pentacyclic Published: 22 November 2016 Triterpene Tatiana M. Souza-Moreira1, Thaís B. Alves1, Karina A. Pinheiro1, Lidiane G. Felippe1, Gustavo M. A. De Lima2, Tatiana F. Watanabe1, Cristina C. Barbosa3, Vânia A. F. F. M. Santos1, Norberto P. Lopes4, Sandro R. Valentini3, Rafael V. C. Guido2, Maysa Furlan1 & Cleslei F. Zanelli3 Among the biologically active triterpenes, friedelin has the most-rearranged structure produced by the oxidosqualene cyclases and is the only one containing a cetonic group. In this study, we cloned and functionally characterized friedelin synthase and one cycloartenol synthase from Maytenus ilicifolia (Celastraceae). The complete coding sequences of these 2 genes were cloned from leaf mRNA, and their functions were characterized by heterologous expression in yeast. The cycloartenol synthase sequence is very similar to other known OSCs of this type (approximately 80% identity), although the M. ilicifolia friedelin synthase amino acid sequence is more related to β-amyrin synthases (65–74% identity), which is similar to the friedelin synthase cloned from Kalanchoe daigremontiana. Multiple sequence alignments demonstrated the presence of a leucine residue two positions upstream of the friedelin synthase Asp-Cys-Thr-Ala-Glu (DCTAE) active site motif, while the vast majority of OSCs identified so far have a valine or isoleucine residue at the same position. The substitution of the leucine residue with valine, threonine or isoleucine in M. ilicifolia friedelin synthase interfered with substrate recognition and lead to the production of different pentacyclic triterpenes. -

Steroidal Triterpenes of Cholesterol Synthesis
Molecules 2013, 18, 4002-4017; doi:10.3390/molecules18044002 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Steroidal Triterpenes of Cholesterol Synthesis Jure Ačimovič and Damjana Rozman * Centre for Functional Genomics and Bio-Chips, Faculty of Medicine, Institute of Biochemistry, University of Ljubljana, Zaloška 4, Ljubljana SI-1000, Slovenia; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +386-1-543-7591; Fax: +386-1-543-7588. Received: 18 February 2013; in revised form: 19 March 2013 / Accepted: 27 March 2013 / Published: 4 April 2013 Abstract: Cholesterol synthesis is a ubiquitous and housekeeping metabolic pathway that leads to cholesterol, an essential structural component of mammalian cell membranes, required for proper membrane permeability and fluidity. The last part of the pathway involves steroidal triterpenes with cholestane ring structures. It starts by conversion of acyclic squalene into lanosterol, the first sterol intermediate of the pathway, followed by production of 20 structurally very similar steroidal triterpene molecules in over 11 complex enzyme reactions. Due to the structural similarities of sterol intermediates and the broad substrate specificity of the enzymes involved (especially sterol-Δ24-reductase; DHCR24) the exact sequence of the reactions between lanosterol and cholesterol remains undefined. This article reviews all hitherto known structures of post-squalene steroidal triterpenes of cholesterol synthesis, their biological roles and the enzymes responsible for their synthesis. Furthermore, it summarises kinetic parameters of enzymes (Vmax and Km) and sterol intermediate concentrations from various tissues. Due to the complexity of the post-squalene cholesterol synthesis pathway, future studies will require a comprehensive meta-analysis of the pathway to elucidate the exact reaction sequence in different tissues, physiological or disease conditions. -

Expression of Selected Genes Involved in Steroidogenesis in the Course of Enucleation-Induced Rat Adrenal Regeneration
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 33: 613-623, 2014 Expression of selected genes involved in steroidogenesis in the course of enucleation-induced rat adrenal regeneration MARIANNA TYCZEWSKA, MARCIN RUCINSKI, AGNIESZKA ZIOLKOWSKA, MARCIN TREJTER, MARTA SZYSZKA and LUDWIK K. MALENDOWICZ Department of Histology and Embryology, Poznan University of Medical Sciences, Poznan, Poland Received October 28, 2013; Accepted December 6, 2013 DOI: 10.3892/ijmm.2013.1599 Abstract. The enucleation-induced (EI) rapid proliferation however, throughout the entire experimental period, there were of adrenocortical cells is followed by their differentiation, no statistically significant differences observed. After the initial the degree of which may be characterized by the expression decrease in steroidogenic factor 1 (Sf-1) mRNA levels observed of genes directly and indirectly involved in steroid hormone on the 1st day of the experiment, a marked upregulation in its biosynthesis. In this study, out of 30,000 transcripts of genes expression was observed from there on. Data from the current identified by means of Affymetrix Rat Gene 1.1 ST Array, we study strongly suggest the role of Fabp6, Lipe and Soat1 in aimed to select genes (either up- or downregulated) involved supplying substrates of regenerating adrenocortical cells for in steroidogenesis in the course of enucleation-induced adrenal steroid synthesis. Our results indicate that during the first days regeneration. On day 1, we found 32 genes with altered of adrenal regeneration, intense synthesis of cholesterol may expression levels, 15 were upregulated and 17 were down- occur, which is then followed by its conversion into cholesteryl regulated [i.e., 3β-hydroxysteroid dehydrogenase (Hsd3β), esters. -
Figures Intermediary Ontogeny.Pptx
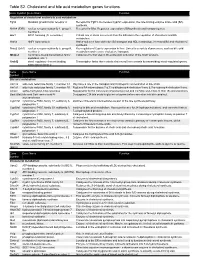
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi. -

Product Data Sheet
Inhibitors Product Data Sheet Ro 48-8071 fumarate • Agonists Cat. No.: HY-18630A CAS No.: 189197-69-1 Molecular Formula: C₂₇H₃₁BrFNO₆ • Molecular Weight: 564.44 Screening Libraries Target: Others Pathway: Others Storage: Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro H2O : 100 mg/mL (177.17 mM; Need ultrasonic) DMSO : ≥ 55 mg/mL (97.44 mM) * "≥" means soluble, but saturation unknown. Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 1.7717 mL 8.8583 mL 17.7167 mL Stock Solutions 5 mM 0.3543 mL 1.7717 mL 3.5433 mL 10 mM 0.1772 mL 0.8858 mL 1.7717 mL Please refer to the solubility information to select the appropriate solvent. In Vivo 1. Add each solvent one by one: 10% DMSO >> 40% PEG300 >> 5% Tween-80 >> 45% saline Solubility: ≥ 2.5 mg/mL (4.43 mM); Clear solution 2. Add each solvent one by one: 10% DMSO >> 90% (20% SBE-β-CD in saline) Solubility: ≥ 2.5 mg/mL (4.43 mM); Clear solution 3. Add each solvent one by one: 10% DMSO >> 90% corn oil Solubility: ≥ 2.5 mg/mL (4.43 mM); Clear solution BIOLOGICAL ACTIVITY Description Ro 48-8071 fumarate is an inhibitor of OSC (Oxidosqualene cyclase) with IC50 of appr 6.5 nM. IC₅₀ & Target IC50: appr 6.5 nM (Oxidosqualene cyclase)[1] [1] In Vitro In HepG2 cells, Ro 48-8071 reduces cholesterol synthesis dose dependently with an IC50 value of appr 1.5 nM . -

SUPPLEMENTARY MATERIAL Supplementary Table 1
Predicting Drug Promiscuity Using Spherical Harmonic Surface The Open Conference Proceedings Journal, 2011, Volume 2 i SUPPLEMENTARY MATERIAL Supplementary Table 1. MDDR Target Annotations Used in the Promiscuity Predictions TARGET_KEY EXTERNAL_ID 1001 prostaglandin-endoperoxide synthase 1002 cyclooxygenase 1 1003 cyclooxygenase 2 1046 ribonucleoside-diphosphate reductase 1112 thymidylate synthase 1209 phosphoribosylglycinamide formyltransferase 1609 purine-nucleoside phosphorylase 1611 uridine phosphorylase 1637 nad adp-ribosyltransferase 1657 transferring alkyl or aryl groups, other than methyl groups 1798 thymidine kinase 1814 protein kinase 1843 1-phosphatidylinositol 4-kinase 1883 protein-tyrosine kinase 1973 nucleotidyltransferase 2018 rna-directed dna polymerase 2121 phospholipase a2 2124 acetylcholinesterase 2282 phosphoric diester hydrolase 2283 phosphodiesterase i 2309 3',5'-cyclic-nucleotide phosphodiesterase 2310 3',5'-cyclic-gmp phosphodiesterase 2316 steryl-sulfatase 2414 alpha-glucosidase 2565 adenosylhomocysteinase 2576 peptidase 25 aldehyde reductase 2613 dipeptidyl-peptidase iv 2619 peptidyl-dipeptidase a 2624 serine-type d-ala-d-ala carboxypeptidase 2659 serine endopeptidase 2663 trypsin 2664 thrombin 2665 coagulation factor xa 2672 coagulation factor viia 2695 tryptase 2742 cathepsin b 2750 cathepsin l 2765 caspase-1 2784 hiv-1 retropepsin 2811 metalloendopeptidase 2822 stromelysin 1 286 xanthine oxidase 2978 beta-lactamase 3014 adenosine deaminase ii The Open Conference Proceedings Journal, 2011, Volume 2 Pérez-Nueno -

Oxidosqualene Cyclase Inhibitors As Antimicrobial Agents
4240 J. Med. Chem. 2003, 46, 4240-4243 Oxidosqualene Cyclase Inhibitors as Scheme 1. Cyclization of Oxidosqualene Antimicrobial Agents Jerald C. Hinshaw,‡ Dae-Yeon Suh,‡ Philippe Garnier,‡ Frederick S. Buckner,§ Richard T. Eastman,§ Seiichi P. T. Matsuda,⊥ ⊥ | Bridget M. Joubert, Isabelle Coppens, 3-5 Keith A. Joiner,| Salim Merali,† humans. Oxidosqualene cyclases (OSCs) are impor- 6 Theodore E. Nash,# and Glenn D. Prestwich*,‡ tant enzymes in triterpenoid biosynthesis. These en- zymes catalyze the cyclization of (3S)-2,3-oxidosqualene Department of Medicinal Chemistry, to lanosterol in fungi, mammals, and some protists and University of Utah, 419 Wakara Way, Suite 205, to cycloartenol, as well as several other pentacyclic Salt Lake City, Utah 84108-1257, Department of Medicine, triterpenes in plants1 (Scheme 1). University of Washington, Box 357185, Seattle, Washington, 98195, Department of Chemistry Recent studies have revealed that a number of human and Department of Biochemistry and Cell Biology, pathogenic microorganisms synthesize sterols. These Rice University, 6100 South Main Street, include Pneumocystis carinii (pneumonia),7 Trypano- Houston, Texas 77005, Infectious Disease Section, soma brucei (African sleeping sickness),8 Trypanosoma Department of Internal Medicine, Yale University School of cruzi (Chagas disease),9 and Leishmania sp. (leish- Medicine, New Haven, Connecticut 06520-8022, 10-12 Department of Medical and Molecular Parasitology, maniasis). Sterol biosynthetic routes have been 13,14 New York University School of Medicine, examined as drug targets against Trypanosome and 341 East 25th Street, New York, New York 10010, and Pneumocystis15 organisms. Laboratory of Parasitic Diseases, National Institutes of Because hypercholesterolemia is a major risk factor Health, Bethesda, Maryland 20892 for the development of atherosclerosis in humans, Received June 9, 2003 considerable research and development have been di- rected toward the inhibition of pathways in cholesterol biosynthesis. -

The Use of Mutants and Inhibitors to Study Sterol Biosynthesis in Plants
bioRxiv preprint doi: https://doi.org/10.1101/784272; this version posted September 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Title page 2 Title: The use of mutants and inhibitors to study sterol 3 biosynthesis in plants 4 5 Authors: Kjell De Vriese1,2, Jacob Pollier1,2,3, Alain Goossens1,2, Tom Beeckman1,2, Steffen 6 Vanneste1,2,4,* 7 Affiliations: 8 1: Department of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 71, 9052 Ghent, 9 Belgium 10 2: VIB Center for Plant Systems Biology, VIB, Technologiepark 71, 9052 Ghent, Belgium 11 3: VIB Metabolomics Core, Technologiepark 71, 9052 Ghent, Belgium 12 4: Lab of Plant Growth Analysis, Ghent University Global Campus, Songdomunhwa-Ro, 119, Yeonsu-gu, Incheon 13 21985, Republic of Korea 14 15 e-mails: 16 K.D.V: [email protected] 17 J.P: [email protected] 18 A.G. [email protected] 19 T.B. [email protected] 20 S.V. [email protected] 21 22 *Corresponding author 23 Tel: +32 9 33 13844 24 Date of submission: sept 26th 2019 25 Number of Figures:3 in colour 26 Word count: 6126 27 28 1 bioRxiv preprint doi: https://doi.org/10.1101/784272; this version posted September 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Oxygen-Mediated Regulation of Cholesterol Synthesis
OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE APPROVED BY SUPERVISORY COMMITTEE Russell DeBose-Boyd, Ph.D. Richard Bruick, Ph.D. Michael Brown, M.D. Zhijian Chen, Ph.D. George DeMartino, Ph.D. To my family For their love and support OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE by ANDREW TUAN DUC NGUYEN DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas May, 2009 Copyright by Andrew Tuan Duc Nguyen, 2009 All Rights Reserved OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE Andrew Tuan Duc Nguyen, Ph.D. The University of Texas Southwestern Medical Center at Dallas, 2009 Supervising Professor: Russell A. DeBose-Boyd, Ph.D. Endoplasmic reticulum-associated degradation of the enzyme 3-hydroxy-3- methylglutaryl CoA reductase represents one mechanism by which cholesterol synthesis is controlled in mammalian cells. The key reaction in this degradation is binding of reductase to Insig proteins in the endoplasmic reticulum, which is stimulated by the methylated cholesterol precursors lanosterol and 24,25-dihydrolanosterol. Conversion of these sterols to cholesterol requires the removal of three methyl groups, which consumes v nine molecules of O2. Here, we report that oxygen deprivation (hypoxia) slows the rate of demethylation of lanosterol and its reduced metabolite 24,25-dihydrolanosterol, causing both sterols to accumulate in cells.