Proquest Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Information to Users
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter &ce, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afreet reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back o f the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6” x 9” black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. UMI A Bell & Howell Io5)nnaüon Company 300 North Zed) Road, Ann Arbor MI 48106-1346 USA 313/761-4700 800/521-0600 LIQUID CRYSTAL AND HYDROPHILIC GEL MEDIA FOR IONTOPHORESIS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Manesh A. -

WO 2018/035147 Al 22 February 2018 (22.02.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/035147 Al 22 February 2018 (22.02.2018) W !P O PCT (51) International Patent Classification: (74) Agent: WORD, Michael, J.; Mayer Brown LLP, P.O. Box G06F 17/00 (2006.01) 2828, Chicago, IL 60690-2828 (US). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/US20 17/046997 kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, (22) International Filing Date: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, 15 August 2017 (15.08.2017) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (25) Filing Language: English HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (26) Publication Langi English MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (30) Priority Data: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, 62/375,256 15 August 2016 (15.08.2016) US SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 62/375,192 15 August 2016 (15.08.2016) US TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. 62/416,972 03 November 2016 (03 .11.20 16) US (84) Designated States (unless otherwise indicated, for every 62/427,919 30 November 2016 (30. -

Pain Relief for the Peripheral Venous Cannulation Of
Bond et al. BMC Anesthesiology (2016) 16:81 DOI 10.1186/s12871-016-0252-8 RESEARCH ARTICLE Open Access First do no harm: pain relief for the peripheral venous cannulation of adults, a systematic review and network meta- analysis Mary Bond1, Louise Crathorne1* , Jaime Peters1, Helen Coelho1, Marcela Haasova1, Chris Cooper1, Quentin Milner2, Vicki Shawyer3, Christopher Hyde1 and Roy Powell4 Abstract Background: Peripheral venous cannulation is an everyday practice in hospitals, which many adults find painful. However, anaesthesia for cannulation is usually only offered to children. Inadequate pain relief is not only unpleasant for patients but may cause anxiety about further treatment and deter patients from seeking medical care in the future. The aim of this study is to discover the most effective local anaesthetic for adult peripheral venous cannulation and to find out how the pain of local anaesthetic application compares with that of unattenuated cannulation. Methods: These aims are addressed through a systematic review, network meta-analysis and random-effects meta-analysis. Searching covered 12 databases including MEDLINE and EMBASE from 1990 to August 2015. The main included study design was RCTs. The primary outcome measure is self-reported pain, measured on a 100 mm visual analogue scale. Results: The systematic review found 37 includable studies, 27 of which were suitable for network meta-analysis and two for random-effects meta-analysis. The results of the network meta-analysis indicate that none of the 17 anaesthetic considered had a very high probability of being the most effective when compared to each other; 2 % lidocaine had the highest probability (44 %). -

204200Orig1s000 204200Orig2s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 204200Orig1s000 204200Orig2s000 MEDICAL REVIEW(S) CLINICAL REVIEW Application Type NDA Application Number(s) 204-200 and 204-640 Priority or Standard Standard Submit Date(s) March 7, 2012 Received Date(s) March 7, 2012 PDUFA Goal Date January 7, 2013 Division / Office DPARP/OND Reviewer Name(s) Peter Starke, MD Review Completion Date October 29, 2012 Established Name Epinephrine injection, USP, 1mg/mL (Proposed) Trade Name Adrenalin® Therapeutic Class Catecholamine Applicant JHP Pharmaceuticals, LLC Formulation(s) Solution for injection Proposed Dosing Hypersensitivity reactions: IM or SC Regimen injection(b) (4) [Ophthalmic use: Topical irrigation or intraocular bolus injection] Proposed Indication(s) Hypersensitivity reactions: severe acute anaphylactic reactions, (b) (4) [Ophthalmic use: induction and maintenance of mydriasis during cataract surgery] Intended Population(s) Hypersensitivity reactions: No age restrictions [Ophthalmic use: No age restrictions] Reference ID: 3209828 Clinical Review ● Peter Starke, MD NDA 204-200 ● Adrenalin® (epinephrine) MEDICAL OFFICER REVIEW Division of Pulmonary, Allergy and Rheumatology Products (DPARP) SUBMISSIONS REVIEWED IN THIS DOCUMENT Document Date CDER Stamp Date Submission Comments March 7, 2012 March 7, 2012 SD-1, eCTD-0000 New NDA submission April 9, 2012 April 9, 2012 SD-2, eCTD-0001 PREA waiver request – Anaphylaxis April 9, 2012 April 9, 2012 SD-3, eCTD-0002 PREA waiver request – Mydriasis April 18, 2012 April 18, 2012 SD-4, eCTD-0003 Request for proprietary name review Sept 5, 2012 Sept 6, 2012 SD-22, eCTD-0021 Draft Ophthalmic Labeling Oct 22, 2012 Oct 22, 2012 SD-27, eCDT-0026 Certification that EpiPen is the reference for the 505(b)(2) application RECOMMENDED REGULATORY ACTION NDA/SUPPLEMENTS: X APPROVAL COMPLETE RESPONSE OTHER ACTION: 2 Reference ID: 3209828 Clinical Review ● Peter Starke, MD NDA 204-200 ● Adrenalin® (epinephrine) Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT ........................................ -

Code of Federal Regulations GPO Access
4±28±97 Monday Vol. 62 No. 81 April 28, 1997 Pages 22873±23124 Briefings on how to use the Federal Register For information on briefings in Washington, DC, see announcement on the inside cover of this issue. For information on briefings in Kansas City and Independence, MO, Long Beach and San Francisco, CA, and Anchorage, AK, see the announcement in the Reader Aids. Now Available Online Code of Federal Regulations via GPO Access (Selected Volumes) Free, easy, online access to selected Code of Federal Regulations (CFR) volumes is now available via GPO Access, a service of the United States Government Printing Office (GPO). CFR titles will be added to GPO Access incrementally throughout calendar years 1996 and 1997 until a complete set is available. GPO is taking steps so that the online and printed versions of the CFR will be released concurrently. The CFR and Federal Register on GPO Access, are the official online editions authorized by the Administrative Committee of the Federal Register. New titles and/or volumes will be added to this online service as they become available. http://www.access.gpo.gov/nara/cfr For additional information on GPO Access products, services and access methods, see page II or contact the GPO Access User Support Team via: ★ Phone: toll-free: 1-888-293-6498 ★ Email: [email protected] federal register 1 II Federal Register / Vol. 62, No. 81 / Monday, April 28, 1997 SUBSCRIPTIONS AND COPIES PUBLIC Subscriptions: Paper or fiche 202±512±1800 Assistance with public subscriptions 512±1806 General online information 202±512±1530; 1±888±293±6498 FEDERAL REGISTER Published daily, Monday through Friday, Single copies/back copies: (not published on Saturdays, Sundays, or on official holidays), by the Office of the Federal Register, National Archives and Paper or fiche 512±1800 Records Administration, Washington, DC 20408, under the Federal Assistance with public single copies 512±1803 Register Act (49 Stat. -

United States Patent (10) Patent No.: US 9,463,201 B2 Alster Et Al
USOO94632O1B2 (12) United States Patent (10) Patent No.: US 9,463,201 B2 Alster et al. (45) Date of Patent: Oct. 11, 2016 (54) COMPOSITIONS AND METHODS FOR THE 2010/02851.55 A1 11, 2010 Gilbard et al. TREATMENT OF MEBOMAN GLAND 2011/0022O10 A1 1/2011 Grenon et al. 2011/0059925 A1 3/2011 Donnenfeld DYSFUNCTION 2011/O124725 A1* 5, 2011 Maskin ................ A61K 31, 167 514,529 (71) Applicant: M.G. Therapeutics, Ltd., Tel Aviv (IL) 2011 0130729 A1 6, 2011 Korb et al. 2011 0137214 A1 6, 2011 Korb et al. (72) Inventors: Yair Alster, Tel Aviv (IL); Omer 2011/0294.897 Al 12/2011 Aberg et al. Rafaeli. Udim (IL); K. Angela 2012fOO16275 A1 1/2012 Korb et al. Macf i M o Park, CA (US); 2012/0028929 A1 2/2012 Power et al. aciariane, Menlo Park, s 2012/0093876 Al 4/2012 Ousler, III et al. Cary Reich, Los Gatos, CA (US); 2012/O128763 A1 5, 2012 Maskin Shimon Amselem, Rehovot (IL); 2012/0190661 A1 7/2012 Trogden et al. Doron Friedman, Carme-Yosef (IL) 2012fO226156 A1 9/2012 Grenon et al. 2012fO264681 A1 10, 2012 Braiman-WikSman et al. 2012fO288575 A1 11, 2012 Gilbard et al. (73) Assignee: M.G. THERAPEUTICS LTD, Tel 2013,0053733 A1 2/2013 Korb et al. Aviv (IL) 2013,013 1171 A1 5.2013 Maskin 2013, O184242 A1 7/2013 Eini et al. (*) Notice: Subject to any disclaimer, the term of this 2013/0224272 A1 8/2013 Gao et al. patent is extended or adjusted under 35 3.855. A. -
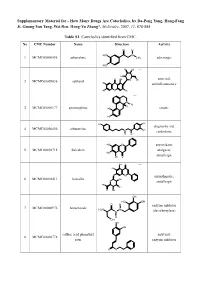
How Many Drugs Are Catecholics, by Da-Peng Yang, Hong-Fang Ji, Guang-Yan Tang, Wei Ren, Hong-Yu Zhang*, Molecules, 2007, 12, 878-884
Supplementary Material for - How Many Drugs Are Catecholics, by Da-Peng Yang, Hong-Fang Ji, Guang-Yan Tang, Wei Ren, Hong-Yu Zhang*, Molecules, 2007, 12, 878-884 Table S1. Catecholics identified from CMC. No CMC Number Name Structure Activity O H HO N 1 MCMC00000595 adrenalone CH3 adrenergic HO HO OH Chiral HO O OH antiviral; 2 MCMC00009836 aphloiol O OH HO antiinflammatory HO O OH Chiral N CH H 3 3 MCMC00000177 apomorphine HO emetic HO HO OH diagnostic aid; 4 MCMC00006456 arbutamine N OH cardiotonic H OH HO O antioxidant; 5 MCMC00010715 baicalein analgesic; HO OH O antiallergic OH O Chiral HO O O antiasthmatic; 6 MCMC00010411 baicalin OH O antiallergic HO OH O OH OH HO OH O H enzyme inhibitor 7 MCMC00000976 benserazide H2N N N (decarboxylase) H OH OH OH caffeic acid phenethyl antiviral; 8 MCMC00006778 ester enzyme inhibitor O O Molecules, 2007, 12, Molecules, 2007, 12, 878-884 2 Table S1. Cont. O OH enzyme inhibitor 9 MCMC00010571 carbidopa CH H N 3 (decarboxylase) HO NH2 OH HO OChiral O O N S N S antibiotic; 10 MCMC00006131 cefetecol H H H2N N N H antibacterial O O HO OH H HO O Chiral HO O 11 MCMC00010530 chf-1301 antiparkinsonian NH CH HO 2 3 OH Chiral HO antiviral; 12 MCMC00000898 cianidol HO O OH antineoplastic HO OH O O OH O O HO phagocyte stimulant; 13 MCMC00007713 cichoric acid O O OH HO antineoplastic HO HO O O HO 14 MCMC00009461 citromycetin O antibiotic HOO CH3 HO OH antispasmodic; 15 MCMC00004237 clofeverine O Cl anticholinergic N H H3C CH3 CH3 16 MCMC00003077 colterol N bronchodilator HO H OH HO HOHO O O 17 MCMC00001729 cynarine O choleretic HO O OH HO OH O OH Molecules, 2007, 12, Molecules, 2007, 12, 878-884 3 Table S1. -
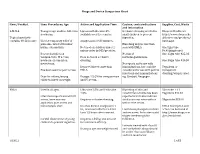
Drugs and Device Comparison Chart
Drugs and Device Comparison Chart Item / Product Uses: Procedures, Age Action and Application Time Caution, contraindications Supplier, Cost, Waste and information L-M-X-4 Youngest age studies- full-term Liposomal Lidocaine 4% Occlusive dressing needed in Eloquest Healthcare newborns. available over the counter. small children to prevent http://www.eloquesthe Topical anesthetic ingestion. althcare.com/products/ contains 4% Lidocaine Use for temporary relief of Analgesia in 20-30 minutes. lmx4.aspx pain associated with minor Blanching may be less than burns, cuts and bits. Not used on children under 2 seen with EMLA. One 5gm tube unless order by MD/provider. $9.90(single use) Research studies on No risk of One 15gm tube $25.03 venipuncture, IV access, Does not need occlusive methemoglobinemia. newborn circumcision, dressing. One 30gm tube $46.30 meatotomy. No reports on its use with Reported faster onset than immunizations, but could be Tegaderm or Has been used for port access. EMLA. considered for use with painful transparent injections and immunizations dressing/wrap is used. Dose for infants/young Dosage: 2.5 GM for venipuncture e.g. Gardasil, Neupogen. children based on weight. and IV access. EMLA Used in all ages. Lidocaine 2.5% and Prilocaine Blanching of skin and 5Gm tube + 12 2.5% constriction of veins has been Tegaderm $44.57 Literature reports use with IV reported but studies indicate access, bone marrow Requires occlusive dressing. similar success rates with or 30gm tube $53.90 aspiration, port access and without the cream. hemodialysis start. Reaches dermal analgesia in 1 EMLA description and hr., maximum effect in 2-3 hrs. -

Review Article Technologies in Transdermal Drug
INTERNATIONAL JOURNAL OF PHARMACEUTICAL AND CHEMICAL SCIENCES ISSN: 22775005 Review Article Technologies in Transdermal Drug Delivery System: A Review Dharmaraj Dnyneshwar Biradar* and Nikita Sanghavi MET’S Institute of Pharmacy, Bandra (west), Mumbai-400 050, India. ABSTRACT Transdermal drug delivery system is the system in which the delivery of the drug occurs through skin. It offers a convenient way to deliver drugs without the drawbacks as in case of standard hypodermic injections relating to issues such as patient acceptability and injection safety. The success of transdermal drug delivery has been severely limited due to the inability of most of the drugs to enter the skin at therapeutically useful rates. The stratum corneum acts as a barrier that limits the penetration of substances through the skin. In recent years various passive and active strategies have emerged to optimise delivery. However passive approach do not significantly improve the permeation of drugs with molecular weight > 500 Da. In contrast active methods, normally involving physical or mechanical methods of enhancing delivery has been shown to be generally superior. The delivery of drugs of differing lipophilicity and molecular weight including proteins, peptides and oligonucletides has been showed to be improved by active methods. This review covers the recent findings in various advanced techniques of enhancement of drug delivery which includes jet injectors, iontophoresis, ultrasound, thermal ablation, biodegradable microneedles. Keywords: Transdermal drug delivery system, iontophoresis, microneedles. INTRODUCTION Transdermal drug delivery systems (TDDS) To increase skin permeability, a number of are defined as self-contained discrete different approaches has been studied, dosage forms which, when applied to intact ranging from chemical/ lipid enhancers to skin, deliver the drug(s), through the skin, electric fields employing iontophoresis and at a controlled rate to systemic circulation. -
Supplement 12
'95 15TH EDITION U.S DEPARTMENT OF HEALTH AND HUMAN$iER\WIC;ES PUBUC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR DRUG EVALUATION ANDHESEAlRCH OFFICE OF MANAGEMENT . IJIVISION OF DRUG INFORMATION RESOURCES SUBSCRIBE NOW! Available in March 1996 New 16th Edition APPROVED DRUG PRODUCTS WITH THERAPEUTIC EQUIVALENCE EVALUATIONS 16lli EDITION 1996 CONTENTS • Prescription Drug Product List .' • OTC Drug Product List • Drug Products with Approval under Section 505 of the Act Administered by the Center for Biologics Evaluation and Research List • Discontinued Drug Product List • USP Monograph Title Additions or Changes • Orphan Drug Product Designations • Drug Products Which Must Demonstrate in vivo Bioavailability Only if Product Fails to Achieve Adequate Dissolution • Biopharmaceutic Guidance Availability • ANDA Suitability Petitions • Patent and Exclusivity Information See Subscription Form Inside Back Cover APPROVED DRUG PRODUCTS with THERAPEUTIC EQUIVALENCE EVALUATIONS 15TH EDITION Cumulative Supplement 12 DECEMBER 1995 CONTENTS PAGE 1.0 INTRODUCTION.............................................. iii 1 .1 How to Use the Cumulative Supplement ........................... iii 1.2 Court Order Affecting Uruguay Round Agreements Act-Extended Patents .... iv 1.3 Products Requiring Revised Labeling for Full Approval . v 1.4 Applicant Name Changes . vi 1.5 Availability of the Publication and Updating Procedures . vii 1.6 Report of Counts for the Prescription Drug Product List. viii 2.0 DRUG PRODUCT LISTS ........................................ 2.1 Prescription Drug Product List ................................. 2.2 OTC Drug Product List ...................................... 68 2.3 Drug Products with Approval under Section 505 of the Act Administered by the Center for Biologics Evaluation and Research List ..... 73 2.4 Orphan Drug Product Designations .............................. 74 2.5 Drug Products Which Must Demonstrate in vivo Bioavailability Only if Product Fails to Achieve Adequate Dissolution ............... -

External Jhpiego PPT Template
Technical Challenges of Male Circumcision: Focus on Anesthesia, Hemostasis, and Closure Kristin Chrouser, MD Jhpiego, Baltimore Critical Underlying Issues Vital Triad: Patient safety Surgical quality Cost effective care Impact of Success/Failure: Potential effects on MC acceptability within communities Potential effects on HIV transmission rates within communities 2 Local Anesthesia for Adult MC Traditional circumcision rites often performed without anesthesia In uncircumcised ethnic groups, use of anesthesia likely to increase acceptability: By “medicalizing” the procedure By preventing significant pain with MC 3 Why consider alternatives to injections? Novice learners have most difficulty with providing and achieving anesthesia using injections Injections are painful Injections have potential complications– anesthetic reactions, hematoma, needlestick injury Potential for unsafe reuse of syringes and needles Cost INJECTIONS ARE A POTENTIAL DETERRENT TO USE OF MC SERVICES 4 Ideal Local Anesthetic Agent Affordable and readily available Stable in tropical climates Easy to administer Minimal risk to the caregiver during administration Administration of anesthetic should be pain free Highly effective (low failure rate) Pain control persists for duration of procedure and into the immediate post-operative period Few complications 5 Options for Local Anesthesia Injection: Ring block Dorsal block Transdermal application: Topical cream (like EMLA) Topical spray Gel sponge + electric current Impregnated patch + heat -

WO 2017/027673 Al 16 February 2017 (16.02.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/027673 Al 16 February 2017 (16.02.2017) P O P C T (51) International Patent Classification: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61J 7/04 (2006.01) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (21) International Application Number: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PCT/US20 16/046491 PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (22) International Filing Date: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 11 August 2016 ( 11.08.2016) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (30) Priority Data: TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, 62/203,638 11 August 2015 ( 11.08.2015) TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, 62/252,966 November 2015 (09. 11.2015) DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, (72) Inventor; and SM, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, (71) Applicant : VALENTINE, Edmund, L.