Charge-Transfer and Impulsive Electronic-To-Vibrational Energy Conversion in Ferricyanide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent 19 11 Patent Number: 5,360,712 Olm Et Al
D US005360712A United States Patent 19 11 Patent Number: 5,360,712 Olm et al. 45 Date of Patent: Nov. 1, 1994 54 INTERNALLY DOPED SILVER HALIDE 4,981,781 1/1991 McDugle et al. ................... 430/605 EMULSIONS AND PROCESSES FOR THER 5,037,732 8/1991 McDugle et al. ................... 430/567 ARA 5,132,203 7/1992 Bell et al. ............. ... 430/567 PREP TION 5,268,264 12/1993 Marchetti et al. .................. 430/605 75) Inventors: McDugle;Myra T. Olm, Sherrin Webster; A. Puckett, Woodrow both G.of FOREIGN PATENT DOCUMENTS Rochester; Traci Y. Kuromoto, West 513748A1 11/1992 European Pat. Off. ... GO3C 7/392 Henrietta; Raymond S. Eachus, Rochester; Eric L. Bell, Wesbter; OTHER PUBLICATIONS Robert D. Wilson, Rochester, all of Research Disclosure, vol. 176, Dec. 1978, Item 17643, N.Y. Section I, subsection A. Research Disclosure, vol. 308, Dec. 1989, Item 308119, 73) Assignee: Eastman Kodak Company, Section, I, subsection D. Rochester, N.Y. 21 Appl. No.: 91,148 Primary Examiner-Janet C. Baxter J. v. V. 19 Attorney, Agent, or Firm-Carl O. Thomas 51) Int. Cl................................................. GC/ A process is disclosed of preparing a radiation sensitive 52 U.S.C. .................................... 430/567; 430/569; silver halide emulsion comprising reacting silver and 430/604; 430/605 halide ions in a dispersing medium in the presence of a 58 Field of Search ................ 430/567, 569, 604, 605 metal hexacoordination or tetracoordination complex (56) References Cited having at least one organic ligand containing a least one carbon-to-carbon bond, at least one carbon-to-hydro U.S. -
![Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009)](https://docslib.b-cdn.net/cover/8644/determination-of-the-activation-parameters-of-reaction-between-fe-cn6-and-k-co-hedta-no2-2009-998644.webp)
Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009)
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 12-2009 Determination of the Activation Parameters of -4 Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]. Sammy Eni Eni East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Organic Chemistry Commons Recommended Citation -4 Eni Eni, Sammy, "Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009). Electronic Theses and Dissertations. Paper 1798. https://dc.etsu.edu/etd/1798 This Thesis - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. -4 The Determination of the Activation Parameters of Reaction Between [Fe(CN)6] and K[Co(HEDTA)NO2] ________________________________ A thesis presented to the faculty of the Department of Chemistry East Tennessee State University in Partial Fulfillment of the requirements for the degree Master of Science in Chemistry ________________________________ by Sammy Eni Eni December 2009 ________________________________ Dr. Jeffrey Wardeska, Chair Dr. Ningfeng Zhao Dr. Yu-Lin Jiang -4 Keywords: Activation parameters, [Fe(CN)6] , K[Co(HEDTA)NO2]. ABSTRACT -4 Determination of the Activation Parameters of Reaction Between [Fe(CN)6] and K[Co(HEDTA)NO2] by Sammy Eni Eni -4 The kinetics of the oxidation of [Fe(CN)6] by K[Co(HEDTA)NO2] was studied in order to get the mechanism and the activation parameters of the reaction. -

Reversibility of Ferri-/Ferrocyanide Redox During Operando Soft X-Ray Spectroscopy
Reversibility of Ferri-/Ferrocyanide Redox during Operando Soft X-ray Spectroscopy The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Risch, Marcel, Kelsey A. Stoerzinger, Tom Z. Regier, Derek Peak, Sayed Youssef Sayed, and Yang Shao-Horn. “Reversibility of Ferri-/ Ferrocyanide Redox During Operando Soft X-Ray Spectroscopy.” The Journal of Physical Chemistry C 119, no. 33 (August 20, 2015): 18903–18910. As Published http://dx.doi.org/10.1021/acs.jpcc.5b04609 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/109590 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reversibility of Ferri-Ferrocyanide Redox During Operando Soft X-Ray Spectroscopy ┴ Marcel Risch,†* Kelsey A. Stoerzinger,§ Tom Z. Regier,‡ Derek Peak,º Sayed Youssef Sayed,† Yang Shao-Horn†§||* †Research Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA, USA 02139 §Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA 02139 ||Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA 02139 ‡Canadian Light Source, Inc., Saskatoon, SK, Canada S7N 2V3 ºDepartment of Soil Sciences, University of Saskatchewan, Saskatoon, SK, Canada S7N 5A8 KEYWORDS. Electrochemistry; In-situ; redox shuttle; iron cyanide; X-ray absorption; radiolysis; radiation damage. ABSTRACT The ferri-ferrocyanide redox couple is ubiquitous in many fields of physical chemistry. We studied its photochemical response to intense synchrotron radiation by in-situ X-Ray absorption spectroscopy. -

Investigation of Novel Nanoparticles of Gallium Ferricyanide and Gallium Lawsonate As Potential Anticancer Agents, and Nanoparti
Investigation of Novel Nanoparticles of Gallium Ferricyanide and Gallium Lawsonate as Potential Anticancer Agents, and Nanoparticles of Novel Bismuth Tetrathiotungstate as Promising CT Contrast Agent A Thesis submitted to Kent State University In partial fulfillment of the requirements for the degree of Master of Science Liu Yang August 2014 Thesis written by Liu Yang B.S. Kent State University, 2013 M.S. Kent State University, 2014 Approved by ___________________________________, Advisor, Committee member Dr. Songping Huang ___________________________________, Committee member Dr. Scott Bunge ___________________________________, Committee member Dr. Mietek Jaroniec Accepted by ___________________________________, Chair, Department of Chemistry Dr. Michael Tubergen ___________________________________, Dean, College of Arts and Sciences Dr. James L. Blank ii Table of Contents List of Figures..…………………………………………………………………........vii Acknowledgements ……………………………………………………………….….xi Chapter 1: Summary, Materials and Methods …..……………………………………1 1.1 Materials ………………………………………………………………….3 1.1.1 carboxymethyl reduced polysaccharide (CMRD) preparation….3 1.2 Methods …………………………………………………………………4 1.2.1 Atomic absorption spectroscopy (AA) …………………………4 1.2.2 Acid base treating method ……………………………………...4 1.2.3 Cell viability study ……………………………………………...5 i) MTT assay…………………………………………………..5 ii) Trypan blue assay ………………………………………….6 1.2.4 Dialysis …………………………………………………………6 1.2.5 Elementary analysis …………………………………………….7 1.2.6 Lyophilization …………………………………………………..7 iii 1.2.7 -
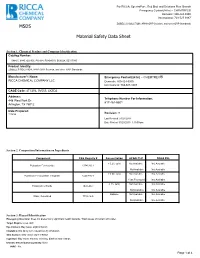
MSDS Material Safety Data Sheet
For RICCA, SpectroPure, Red Bird, and Solutions Plus Brands Emergency Contact(24 hr) -- CHEMTREC® Domestic: 800-424-9300 International: 703-527-3887 ZOBELL'S SOLUTION, APHA ORP Solution, and other ORP Standards MSDS Material Safety Data Sheet Section 1: Chemical Product and Company Identification Catalog Number: 5464.5, 9880, OX-905, PX-918, R5464510, S0932A, SZ117300 Product Identity: ZOBELL'S SOLUTION, APHA ORP Solution, and other ORP Standards Manufacturer's Name: Emergency Contact(24 hr) -- CHEMTREC® RICCA CHEMICAL COMPANY LLC Domestic: 800-424-9300 International: 703-527-3887 CAGE Code: 4TCW6, 0V553, 4XZQ2 Address: Telephone Number For Information: 448 West Fork Dr 817-461-5601 Arlington, TX 76012 Date Prepared: 11/9/98 Revision: 9 Last Revised: 01/24/2014 Date Printed: 03/25/2015 1:19:09 pm Section 2. Composition/Information on Ingredients Component CAS Registry # Concentration ACGIH TLV OSHA PEL < 0.2% (w/v) Not Available Not Available Potassium Ferricyanide 13746-66-2 Not Available Not Available < 0.5% (w/v) Not Available Not Available Potassium Ferrocyanide Trihydrate 14459-95-1 1 (as Fe) mg/m3 Not Available < 1% (w/v) Not Available Not Available Potassium Chloride 7447-40-7 Not Available Not Available Balance Not Available Not Available Water, Deionized 7732-18-5 Not Available Not Available Section 3: Hazard Identification Emergency Overview: Does not present any significant health hazards. Wash areas of contact with water. Target Organs: eyes, skin Eye Contact: May cause slight irritation. Inhalation: Not likely to be hazardous by inhalation. Skin Contact: May cause slight irritation. Ingestion: May cause nausea, vomiting, diarrhea and cramps. -

The Light Triggered Dissolution of Gold Wires Using Potassium Ferrocyanide T Solutions Enables Cumulative Illumination Sensing ⁎ Weida D
Sensors & Actuators: B. Chemical 282 (2019) 52–59 Contents lists available at ScienceDirect Sensors and Actuators B: Chemical journal homepage: www.elsevier.com/locate/snb The light triggered dissolution of gold wires using potassium ferrocyanide T solutions enables cumulative illumination sensing ⁎ Weida D. Chena, Seung-Kyun Kangb, Wendelin J. Starka, John A. Rogersc,d,e, Robert N. Grassa, a Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland b Department of Bio and Brain Engineering, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 334141, Republic of Korea c Departments of Materials Science and Engineering, Biomedical Engineering, Neurological Surgery, Chemistry, Mechanical Engineering, Electrical Engineering and Computer Science, Northwestern University, Evanston, IL 60208, USA d Center for Bio-Integrated Electronics, Northwestern University, Evanston, IL 60208, USA e Simpson Querrey Institute for Nano/Biotechnology, Northwestern University, Evanston, IL 60208, USA ARTICLE INFO ABSTRACT Keywords: Electronic systems with on-demand dissolution or destruction capabilities offer unusual opportunities in hard- Photochemistry ware-oriented security devices, advanced military spying and controlled biological treatment. Here, the dis- Cyanide solution chemistry of gold, generally known as inert metal, in potassium ferricyanide and potassium ferrocya- Sensor nide solutions has been investigated upon light exposure. While a pure aqueous solution of potassium Conductors ferricyanide–K3[Fe(CN)6] does not dissolve gold, an aqueous solution of potassium ferrocyanide–K4[Fe(CN)6] Diffusion limitation irradiated with ambient light is able to completely dissolve a gold electrode within several minutes. Photo Devices activation and dissolution kinetics were assessed at different initial pH values, light irradiation intensities and ferrocyanide concentrations. -

Long-Term Ferrocyanide Application Via Deicing Salts Promotes the Establishment of Actinomycetales Assimilating Ferrocyanide-Derived Carbon in Soil
bs_bs_banner Long-term ferrocyanide application via deicing salts promotes the establishment of Actinomycetales assimilating ferrocyanide-derived carbon in soil Silvia Gschwendtner,1 Tim Mansfeldt,2 soils, belonging mostly to Actinomycetales Susanne Kublik,1 Evangelia Touliari,1 (Kineosporia, Mycobacterium, Micromonosporaceae). Franz Buegger3 and Michael Schloter1,* In the soil without pre-exposition, bacteria belonging 1Research Unit Environmental Genomics, Helmholtz to Acidobacteria (Gp3, Gp4, Gp6), Gemmatimonade- Zentrum Munchen,€ German Research Center for tes (Gemmatimonas)andGammaproteobacteria Environmental Health (GmbH), Ingolstadter€ Landstraße 1, (Thermomonas, Xanthomonadaceae)usedferro- Neuherberg 85764, Germany. cyanide as C source but not the present Actinomyc- 2Department Geowissenschaften, Bodengeographie/ etales. This indicated that (i) various bacteria are able Bodenkunde, Universitat€ zu Koln,€ Albertus-Magnus- to assimilate ferrocyanide-derived C and (ii) long-term Platz, Koln€ 50923, Germany. exposition to ferrocyanide applied with deicing salts 3Institute of Biochemical Plant Pathology, Helmholtz leads to Actinomycetales outcompeting other microor- Zentrum Munchen,€ German Research Center for ganisms for the use of ferrocyanide as C source. Environmental Health (GmbH), Ingolstadter€ Landstraße 1, Neuherberg 85764, Germany. Introduction Cyanide is produced by various organisms including Summary bacteria, algae, fungi and higher plants as defence Cyanides are highly toxic and produced by various mechanism or offensive strategy (Møller and Seigler, microorganisms as defence strategy or to increase 1998; Gallagher and Manoil, 2001; Zagrobelny et al., their competitiveness. As degradation is the most 2008) and consequently occurs naturally at low levels in efficient way of detoxification, some microbes devel- the environment. However, it is highly toxic for living oped the capability to use cyanides as carbon and organisms by complexing metalloproteins, e.g. -

Problems 14, 15, 16, 19, 22, 24, and 25 in Dekock and Gray, Pages 411–414. 14. “All Octahedral Complexes of V3+ Have The
Problems 14, 15, 16, 19, 22, 24, and 25 in DeKock and Gray, pages 411–414. 14. “All octahedral complexes of V3+ have the same number of unpaired electrons, no matter what the nature of the ligand. Why is this so?” V3+ has a d2 electron configuration; in an octahedral ligand environment, regardless of the type of ligand interactions, the lowest energy orbitals are the triply degenerate 2 t2g orbitals. A d octahedral metal complex will always have 2 unpaired electrons. For the same reason, octahedral d1, d3, d8, d9, and d10 also only have one possible electronic configuration. 15. “How does the molecular-orbital theory account for the order of ligands in the spectrochemical series.” The spectrochemical series (courtesy of Wikipedia): I− < Br− < S2− < SCN− < Cl− < − − − − 2− − NO3 < N3 < F < OH < C2O4 ≈ H2O < NCS < CH3CN < pyridine < NH3 < − − ethylenediamine < 2,2'-bipyridine < 1,10-phenanthroline < NO2 < PPh3 < CN ≈ CO. This series can be explained by the type of π interactions on the ligands. The ligands with the smallest octahedral splitting will be π-donors (i.e. I-, Br-, etc) who will bond with the t2g orbitals on the metal, raising it in energy, thus decreasing the - t2g eg splitting. Similarly, π-acceptors (i.e. CN , CO, etc) will have the largest octahedral splitting energy as the t2g orbitals on the metal now become bonding and decrease in energy. The ligands in the middle of the series have little π-interaction with the metal and therefore the t2g remain nonbonding. 3- 4- 16. “Explain why Co(CN)6 is extremely stable but Co(CN)6 is not.” 3- 6 6 4- 7 6 1 Co(CN)6 is low spin d , therefore t2g , while Co(CN)6 is low spin d , t2g eg* . -

YSI 3682 Zobell Solution
YSI 3682 Zobell Solution YSI Inc. Chemwatch Hazard Alert Code: 2 Version No: 2.2 Issue Date: 09/27/2018 Safety Data Sheet according to OSHA HazCom Standard (2012) requirements Print Date: 09/27/2018 S.GHS.USA.EN SECTION 1 IDENTIFICATION Product Identifier Product name YSI 3682 Zobell Solution Synonyms 061320, 061321, 061322 Other means of identification Not Available Recommended use of the chemical and restrictions on use Relevant identified uses Calibration of analytical instruments / Reagent. Name, address, and telephone number of the chemical manufacturer, importer, or other responsible party Registered company name YSI Inc. Address 1700/1725 Brannum Ln Yellow Springs OH 45387 United States Telephone (937) 767-7241 Fax Not Available Website www.ysi.com Email [email protected] Emergency phone number Association / Organisation CHEMTREC Emergency telephone numbers (800) 424-9300 Other emergency telephone 011 703-527-3887 numbers SECTION 2 HAZARD(S) IDENTIFICATION Classification of the substance or mixture CHEMWATCH HAZARD RATINGS Min Max Flammability 0 Toxicity 2 0 = Minimum Body Contact 2 1 = Low Reactivity 0 2 = Moderate Note: The hazard category numbers found in GHS classification in section 2 of this 3 = High Chronic 2 SDSs are NOT to be used to fill in the NFPA 704 diamond. Blue = Health Red = 4 = Extreme Fire Yellow = Reactivity White = Special (Oxidizer or water reactive substances) CANADIAN WHMIS SYMBOLS Skin Corrosion/Irritation Category 2, Eye Irritation Category 2A, Germ cell mutagenicity Category 2, Specific target organ toxicity - single exposure Classification Category 3 (respiratory tract irritation), Acute Aquatic Hazard Category 2, Chronic Aquatic Hazard Category 3 Label elements Continued.. -

Treatment of a Silver Dragon for the Removal of Silver Cyanide and Chalconatronite
TREATMENT OF A SILVER DRAGON FOR THE REMOVAL OF SILVER CYANIDE AND CHALCONATRONITE Donna K. Strahan ABSTRACT--Analysis of the corrosion products on a silver dragon suspected to have been treated with cyanide revealed both silver cyanide and chalconatronite. It appears that both of these corrosion products can be formed on silver objects cleaned or replated using a cyanide solution. These corrosion products darken with exposure to light; therefore, they may be mistaken for silver sulfide. In order to determine how to remove the highly poisonous cyanide corrosion product, numerous tests were run. Using a carefully timed procedure the silver cyanide was removed with an aqueous solution of 20% sodium thiosulfate. The chalconatronite was removed mechanically. INTRODUCTION CORROSION FOUND ON a silver Japanese dragon was brought to the attention of the conservators at the Walters Art Gallery because of its unusual bright blue corrosion deposits. Upon examination, a gray corrosion product which turned black with time was also found in association with the blue corrosion. Samples of both corrosion products were analysed. The blue product was identified by X-ray diffraction as chalconatronite (Na2Cu(CO3)2.3H20). The black material was identified by X-ray dif- fraction as silver cyanide (AgCN),1 and the author has previously reported its occur- rence in a preliminary article.2 A spot test for silver cyanide is found in Feigl but has not been tried by the author (see Appendix). This paper will discuss the rarity and formation of chalconatronite on a silver object and the tests and methods used to remove the silver cyanide corrosion. -

Disentangling Transient Charge Density and Metal–Ligand Covalency in Photoexcited Ferricyanide with Femtosecond Resonant Inela
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. Letter Cite This: J. Phys. Chem. Lett. 2018, 9, 3538−3543 pubs.acs.org/JPCL Disentangling Transient Charge Density and Metal−Ligand Covalency in Photoexcited Ferricyanide with Femtosecond Resonant Inelastic Soft X‑ray Scattering † ‡ † § § § ∥ Raphael M. Jay, Jesper Norell, Sebastian Eckert, , Markus Hantschmann, Martin Beye, , § § ⊥ ⊥ ⊥ Brian Kennedy, Wilson Quevedo, William F. Schlotter, Georgi L. Dakovski, Michael P. Minitti, ⊥ ⊥ ● ⊥ # # ◆ Matthias C. Hoffmann, Ankush Mitra, , Stefan P. Moeller, Dennis Nordlund, Wenkai Zhang, , # # ∥ ▲ ∥ Huiyang W. Liang, Kristjan Kunnus, Katharina Kubicek,̌ , Simone A. Techert, ,@ △ ∇ § # ‡ Marcus Lundberg, , Philippe Wernet, Kelly Gaffney, Michael Odelius,*, † § and Alexander Föhlisch*, , † Institut für Physik und Astronomie, Universitaẗ Potsdam, 14476 Potsdam, Germany ‡ Department of Physics, Stockholm University, Albanova University Center, 10691 Stockholm, Sweden § Institute for Methods and Instrumentation for Synchrotron Radiation Research, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, 12489 Berlin, Germany ∥ DESY Photon Science, 22607 Hamburg, Germany ⊥ LCLS, SLAC, Menlo Park, California 94025, United States # PULSE Institute, SLAC, Menlo Park, California 94025, United States @Institute for X-ray Physics, Göttingen University, 37077 Göttingen, Germany △ ̇ Department of Chemistry -

Colorful Iron Complexes Transition Metal Complex Ions SCIENTIFIC
Colorful Iron Complexes Transition Metal Complex Ions SCIENTIFIC Introduction Easily distinguish between solutions of iron(II) and iron(III) ions by performing re-dox reactions between iron’s two oxidation states. Simply add various complex ions to solutions of iron(II) or iron(III)—observe formation of the beautifully- colored Prussian blue precipitate or of the deep blood-red complex, the confirming test for iron(III). Concepts • Complex ions • Oxidation–reduction • Oxidation numbers • Transition metals Materials Iron(III) chloride solution, 0.02 M, FeCl36H2O, 60 mL Colorful Iron Complexes Worksheet Iron(II) sulfate solution, 0.02 M, FeSO47H2O, 60 mL Graduated cylinder, 25-mL Potassium ferricyanide solution, 0.1 M, K3Fe(CN)6, 10 drops Stoppers to fit tubes, 6 Potassium ferrocyanide solution, 0.1 M, K4Fe(CN)63H2O, 10 drops Test tubes, 25 × 150 mm, 6 Potassium thiocyanate solution, 0.1 M, KSCN, 10 drops Test tube rack Water, distilled or deionized, 120 mL Safety Precautions This activity requires the use of hazardous components and/or has the potential for hazardous reactions. Potassium ferricyanide, potassium ferrocyanide, and potassium thiocyanate are dangerous if heated or in contact with concentrated acids since toxic hydrogen cyanide gas may be liberated. Potassium thiocyanate is moderately toxic by ingestion. Potassium ferricyanide, potassium ferrocyanide, and ferrous sulfate are slightly toxic by ingestion. Iron(II) sulfate is corrosive to skin, eyes, and mucous membranes. Iron(III) chloride may be a skin and tissue irritant. Avoid body contact with all chemicals. Wear chemical splash goggles, chemical-resistant gloves, and a chemical-resistant apron. Please review current Material Safety Data Sheets for additional safety, handling, and disposal information.