Table of Contents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, and a FEW CASE STUDIES
Safety Moment TYLER LAB GROUP MEETING 1 Safety Moment TYLER LAB GROUP MEETING 2 Metal Hydrides: Benchtop vs. Box Hydride = :H- Hydrides are powerful Lewis bases and reducing agent ◦ Exothermically form H2 (this should scare you) ◦ Heating leads to faster reactivity ◦ H evolution leads to rapid increase in pressure2 ◦ Uncontrolled reactions easily cause runaway exotherm, class D fire, explosion, and death/unemployment LiAlH is the #1 chemical cause of fatality in chemical4 industry 3 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” LiAlH4 ◦ Insanely irritating (serious safety hazard) ◦ Extremely moisture sensitive (don’t leave out for >2 minutes) ◦ Ethereal mixtures are pyrophoric! DiBuAl-H ◦ Pyrophoric – it will explode upon exposure to oxygen NaEt3BH ◦ Pyrophoric in solution LiH and NaH ◦ Can be handled on the benchtop (not >2 minutes) ◦ Parrafin oil dispersions much safer KH ◦ Pyrophoric if not in a dispersion ◦ Handle with extreme care! † Sirius Black, Harry Potter and the Prisoner of Azkaban 4 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” CaH2 ◦ Very safe to handle on the benchtop Pt-H, Pd-H, Ni-H ◦ All very pyrophoric NaBH4 ◦ Very safe in general Other hydrides ◦ Treat as pyrophoric ◦ Transition metal hydrides vary in hydridic strength ◦ General rule of thumb: if it does hydrogenations, it is probably pyrophoric ◦ If they’re in organics of any kind, they are probably pyrophoric † Sirius Black, Harry Potter and the Prisoner of Azkaban 5 Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Transmetallation Versus Hydride Elimination
DOI: 10.1002/chem.201102678 Transmetallation Versus b-Hydride Elimination: The Role of 1,4- Benzoquinone in Chelation-Controlled Arylation Reactions with Arylboronic Acids Christian Skçld,*[a] Jonatan Kleimark,[b] Alejandro Trejos,[a] Luke R. Odell,[a] Sten O. Nilsson Lill,[b] Per-Ola Norrby,[b] and Mats Larhed[a] Abstract: The formation of an atypical, um, which is necessary to complete the complex. The association of BQ lowers saturated, diarylated, Heck/Suzuki, catalytic cycle, this electron-deficient the free-energy barrier for transmetal- domino product produced under oxida- alkene opens up a low-energy reaction lation of the s-alkyl complex to create tive Heck reaction conditions, employ- pathway from the post-insertion s-alkyl a pathway that is energetically lower ing arylboronic acids and a chelating than the oxidative Heck reaction path- vinyl ether, has been investigated by way. Furthermore, the calculations Keywords: arylation · CÀC cou- DFT calculations. The calculations showed that the reaction is made pling · density functional calcula- highlight the crucial role of 1,4-benzo- viable by BQ-mediated reductive elimi- tions · elimination · palladium · quinone (BQ) in the reaction. In addi- nation and leads to the saturated di- transmetallation tion to its role as an oxidant of palladi- arylatedACHTUNGRE product. Introduction step of the reaction, it was initially performed by using stoi- ACHTUNGRE chiometric amounts of Pd(OAc)2. More recent develop- The Mizoroki–Heck reaction[1] is a convenient and versatile ments have rendered the reaction catalytic by the re-oxida- method for the formation of carbon–carbon bonds through tion of Pd0 to PdII and today the oxidative Heck reaction is the vinylation or arylation of alkenes.[2] The reaction is per- an established and useful variant of the vinylic substitution formed by Pd0 catalysis in which the active PdII–vinyl or reaction.[4] –aryl intermediate is produced by oxidative addition of aryl One strategy to achieve regio- and stereoselectivity in the halides or pseudo-halides. -
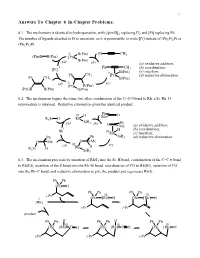
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

The Emergence of Transition Metal-Mediated Hydrothiolation of Unsaturated Carbon-Carbon Bonds: a Mechanistic Outlook Ricardo Castarlenas,* Andrea Di Giuseppe, Jesús J
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital.CSIC Hydrothiolation of Unsaturated Bonds The Emergence of Transition Metal-Mediated Hydrothiolation of Unsaturated Carbon-Carbon Bonds: A Mechanistic Outlook Ricardo Castarlenas,* Andrea Di Giuseppe, Jesús J. Pérez-Torrente, Luis A. Oro* The hydrothiolation of unsaturated carbon-carbon bonds is a practical and atom-economical approach for the incorporation of sulfur into organic frameworks. In recent years, we have witnessed the development of a range of transition metal-based catalytic systems for the control of the regio- and stereoselectivity. This minireview aims to highlight the mechanistic background behind this transformation in order to help for the design of more specific and active organometallic hydrothiolation catalysts. 1. Introduction in-depth understanding of the mechanistic issues, and indeed, it could be useful to other diverse hydroelementation transformations The development of efficient synthetic methods for the such as hydroalkoxylation, hydrophosphination, hydroamination, or incorporation of sulfur into organic frameworks is nowadays an hydroacylation, among others. Our aim in this concise survey is to important task due to the practical applications of this type of analyze the different mechanistic pathways rather than to present a compounds as pharmaceuticals, functional materials, or synthetic comprehensive overview of a research area that has already been reagents.[1-4] In this context, one of the simplest and atom- covered in several recent reviews.[1,5] The sulfa-michael additions economical approach is the direct addition of sulfur and hydrogen mediated by organocatalysts remains beyond the scope of this atoms from thiols to unsaturated carbon-carbon bonds, commonly minireview. -

Insertion Reactions • Oxidative Addition and Substitution Allow Us to Assemble 1E and 2E Ligands on the Metal, Respectively
Insertion Reactions • Oxidative addition and substitution allow us to assemble 1e and 2e ligands on the metal, respectively. • With insertion, and its reverse reaction, elimination, we can now combine and transform these ligands within the coordination sphere, and ultimately expel these transformed ligands to form free organic compounds. • There are two main types of insertion 1) 1,1 insertion in which the metal and the X ligand end up bound to the same (,)(1,1) atom 2) 1,2 insertion in which the metal and the X ligand end up bound to adjacent (1,2) atoms of an L‐type ligand. • The type of insertion observed in any given case depends on the nature of the 2e inserting ligand. • For example: CO gives only 1,1 insertion ethylene gives only 1,2 insertion, in which the M and the X end up on adjacent atoms of what was the 2e X‐type ligand. In general, η1 ligands tend to give 1,1 insertion and η2 ligands give 1,2 insertion • SO2 is the only common ligan d tha t can give bthboth types of itiinsertion; as a ligan d, SO2 can be η1 (S) or η2 (S, O). • In principle, insertion reactions are reversible, but just as we saw for oxidative addition and reductive elimination previously, for many ligands only one of the two possible directions is observed in practice, probably because this direction is strongly favored thermodynamically. •A2e vacant site is generated by 1,1 and 1,2 insertion reactions. • Thissite can be occupidied byanextlternal 2e ligan d and the itiinsertion prodtduct tdtrapped. -

Solventless Migratory-Insertion Reactions of Substituted Cyclopentadienyl Iron Complexes Induced by Electron Donor Ligands
Bull. Chem. Soc. Ethiop. 2009 , 23(3), 399-407. ISSN 1011-3924 Printed in Ethiopia 2009 Chemical Society of Ethiopia SOLVENTLESS MIGRATORY-INSERTION REACTIONS OF SUBSTITUTED CYCLOPENTADIENYL IRON COMPLEXES INDUCED BY ELECTRON DONOR LIGANDS Apollinaire Munyaneza 1, Olalere G. Adeyemi 1, 2 and Neil J. Coville 1* 1Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg, 2050, South Africa 2Department of Chemical Sciences, College of Natural Sciences, Redeemer’s University, Km 46, Lagos/Ibadan Expressway, Redemption City, Nigeria (Received February 24, 2009; revised July 2, 2009) ABSTRABSTRACT.ACT. Reaction between solid (C 5H5)Fe(CO) 2CH 3 and a range of solid phosphine ligands, L (L = PPh 3, P( m-CH 3C6H4)3, P( p-CH 3C6H4)3, P( p-FC 6H4)3, P( p-ClC 6H4)3, PCy 3) occurred in the absence of solvent in the melt phase to give the migratory-insertion products, (C 5H5)Fe(CO)(COCH 3)(L). The reaction was more rapid with small electron withdrawing ligands. Insertion reaction between (RC 5H4)Fe(CO) 2R’ (R = H, CH 3; R’ = CH 3, CH 2Ph) and gaseous ligands such as SO 2 and CO were also studied. The insertion of SO 2 occurred readily for all the substrates investigated, but CO insertion did not occur (< 1 %) using the solventless reaction condition. KEY WORDS: Melt phase, Solventless chemistry, Cyclopentadiene, Iron complexes, Migratory-insertion INTRODUCTION An insertion reaction occurs when an incoming ligand is incorporated into an existing complex without releasing any of the previously coordinated groups [1]. Generally, small molecules such as CO, SO 2, carbenes, etc. -

Computational Aspects of Hydroformylation
RSC Advances REVIEW Computational aspects of hydroformylation Tamas Kegl* Cite this: RSC Adv.,2015,5,4304 ´ ´ The influence of transition metal complexes as catalysts upon the activity and selectivity of hydroformylation reactions has been extensively investigated during the last decades. Nowadays Received 25th October 2014 computational chemistry is an indispensable tool for elucidation of reaction mechanisms and for Accepted 4th December 2014 understanding the various aspects which govern the outcome of catalytic reactions. This review DOI: 10.1039/c4ra13121e attempts to survey the recent literature concerning computational studies on hydroformylation and www.rsc.org/advances theoretical coordination chemistry results related to hydroformylation. 1. Introduction Various transition metal compounds can serve as catalyst precursor for hydroformylation, such as cobalt,2 rhodium,3–6 The hydroformylation or oxo reaction, discovered by Otto platinum,7 ruthenium,8,9 and iridium10 complexes. Very few Roelen1 is the transition metal mediated addition of carbon examples are known about the hydroformylation activity of iron monoxide and dihydrogen to the double bond of an alkene. It is complexes,11,12 although synergistic effect with rhodium one of the most versatile methods for the functionalization of complexes has been also reported.13 C]C bonds and can be considered consequently as a very Hydroformylation is one of the leading industrial process robust synthetic tool. The primary products of the exothermic among homogeneously catalysed reactions producing more reaction are aldehyde isomers. If the carbon atoms of the than 10 million metric tons of oxo chemicals per year.14 Formed double bond are equivalent, only one aldehyde is formed, as aldehydes can be valuable nal products, but oen they are depicted in Scheme 1(a). -

1 IE. Organometallics I. Basic Principles A. Organometallic
I. Basic Principles IE. Organometallics A. Organometallic Mechanisms Oxidation State: The oxidation state of a metal is defined as the charge left on the metal after all ligands have been removed in their natural, closed-shell configuration. This is a formalism and not a physical property! d-Electron Configuration: position in the periodic table minus oxidation state. 18-Electron Rule: In mononuclear, diamagnetic complexes, the total number of electrons never exceeds 18 (noble gas configuration). The total number of electrons is equal to the sum of d-electrons plus those contributed by the ligands. 18 electrons = coordinatively saturated < 18 electrons = coordinatively unsaturated. 1 2 Bonding considerations for M-CO: 3 Structure • saturated (18 e-) complexes: - tetracoordinate: Ni(CO)4, Pd(PPh3)4 are tetrahedral - pentacoordinate: Fe(CO)5 is trigonal bipyramidal - hexacoordinate: Cr(CO)6 is octahedral • unsaturated complexes have high dx2-y2; 16e- prefers square planar Basic reaction mechanisms - ligand substitution: M-L + L’ → M-L’ + L can be associative, dissociative, or radical chain. trans-effect: kinetic effect of a ligand on the role of substitution at the position trans to itself in a square or octahedral complex (ground-state weakening of bond). L → M, repels negative charge to trans position. 4 - oxidative addition: - reductive elimination: the major way in which transition metals are used to make C,C- and C,H-bonds! 5 - migratory insertion: - β-elimination/hydrometalation: 6 - olefin metathesis: - transmetalation: R-M + M’-X → R-M’ + M-X 7 Summary of Mechanisms: - ligand substitution - oxidative addition/reductive elimination - migratory insertion/β-elimination (carbo-, hydrometalation) - alkene metathesis - transmetalation IF. -

REVIEW Methyl Complexes of the Transition Metals
REVIEW Methyl Complexes of the Transition Metals Jesús Campos,[b] Joaquín López-Serrano,[a] Riccardo Peloso,[a] and Ernesto Carmona*[a] Dedicated to Prof. Pierre Braunstein in recognition of his contributions to inorganic and organometallic chemistry [a] Title(s), Initial(s), Surname(s) of Author(s) including Corresponding Author(s) Department Institution Address 1 E-mail: [b] Title(s), Initial(s), Surname(s) of Author(s) Department Institution Address 2 Supporting information for this article is given via a link at the end of the document.((Please delete this text if not appropriate)) REVIEW Abstract: Organometallic chemistry can be considered as a wide focusing in the last 5 years. It begins with a brief description of area of knowledge that combines concepts of classic organic synthetic methods, followed by a selection of recently reported chemistry, i.e. based essentially on carbon, with molecular inorganic complexes with terminal and bridging methyl ligands from the chemistry, especially with coordination compounds. Transition metal groups 3 to 11 of the periodic table. A specific section is methyl complexes probably represent the simplest and most dedicated to methyl-bridged species with three-centre two- fundamental way to view how these two major areas of chemistry electron bonds. The review concludes highlighting relevant combine and merge into novel species with intriguing features in reactivity of the methyl group in this class of compounds. terms of reactivity, structure, and bonding. Citing more than 500 bibliographic references, this review aims to offer a concise view of recent advances in the field of transition metal complexes containing Ernesto Carmona (PhD degree, University M—CH3 fragments. -

Insertion of an Alkene Into an Ester: Intramolecular Oxyacylation Reaction of Alkenes Through Acyl CÀO Bond Activation** Giang T
Communications DOI: 10.1002/anie.201005767 CÀO Activation Insertion of an Alkene into an Ester: Intramolecular Oxyacylation Reaction of Alkenes through Acyl CÀO Bond Activation** Giang T. Hoang, Venkata Jaganmohan Reddy, Huy H. K. Nguyen, and Christopher J. Douglas* b-Alkoxyketones are common intermediates in organic syn- thesis. An unusual approach to this class of compounds could be the insertion of a C=C bond into an ester, a potentially atom economical process (Scheme 1). This alkene “oxyacy- Scheme 1. Alkene oxyacylation. Scheme 2. Processes involving acyl CÀO activation. lation” could be an alternative to the aldol reaction.[1] Atom economy and ester manipulation, however, are rarely com- patible: esters usually fragment after reactions with nucleo- approach was akin to stoichiometric acyl CÀO bond activa- philes, or decarbonylate when activated with transition tion strategies[6–8] in which metal chelating groups were metals.[2] In the rare cases when the acyl CÀO bond is used.[6] We employed quinoline as a chelating group based activated and decarbonylation is suppressed, the acyl metal on our previous successes in acyl CÀC bond activation.[9] We alkoxide complexes can undergo additional transforma- designed an intramolecular reaction to avoid problems with tions,[3,4] but only with the expulsion of an alcohol regioselectivity and to increase local concentrations of alkene. (Scheme 2a)[3b–f] or ketone (Scheme 2b).[3a] We are aware of Activation of 1a to I, followed by migratory insertion and one example where acyl CÀO activation provided products reductive elimination would provide 2a, containing a cyclic containing the original atoms: Ohes recent Pd-catalyzed ether with a ketone in a 1,3-relationship and a new fully nitrile insertion into an acyl CÀO bond, followed by substituted carbon center. -

• Oxidative Addition • Reductive Elimination • Migratory Insertion • Β - Hydrogen Elimination AJELIAS L7-S2 Oxidative Addition
AJELIAS L7-S1 Unique reactions in organometallic chemistry • Oxidative Addition • Reductive Elimination • Migratory Insertion • β - Hydrogen Elimination AJELIAS L7-S2 Oxidative addition When addition of ligands is accompanied by oxidation of the metal, it is called an oxidative addition reaction LnM+XY Ln(X)(Y)M dn dn-2 OX state of metal increases by 2 units H H2 oxidative Ph3P PPh3 addition H PPh3 Coordination number increases by 2 units Rh Rh Ph3P Cl Ph3P Cl PPh3 2 new anionic ligands are added to the metal Rh+1 Rh+3 Requirements for oxidative addition • availability of nonbonded electron density on the metal, • two vacant coordination sites on the reacting complex (LnM), that is, the complex must be coordinatively unsaturated, • a metal with stable oxidation states separated by two units; the higher oxidation state must be energetically accessible and stable. AJELIAS L7-S3 Examples of Oxidative addition : Cis or trans ? Cl Cl PPh3 Ir 18E Cl2 Ph3P CO Cl O Cl PPh3 O2 O PPh3 Ir Ir Ph3P CO Ph3P CO 16E Cl Me Me MeI Cl PPh 3 I PPh3 Ir Ir Ph P CO 3 Ph3P CO I Cl Homonuclear systems (H2, Cl2, O2, C2H2) Cis Heteronuclear systems (MeI) Cis or trans AJELIAS L7-S4 An important step in many homogeneous catalytic cycles Hydrogenation of alkenes- Wilkinson catalyst H H2 oxidative Ph3P PPh3 addition H PPh3 Rh Rh Often thefirststepofmechanism Ph3P Cl Ph3P Cl PPh3 Rh+1 Rh+3 Methanol to acetic acid conversion- Cativa process CH3 I CO CH3I I CO Ir Ir I CO I CO Ir+1 Ir+3 I Pd catalyzed Cross coupling of Ar-B(OH)2 and Ar-X – Suzuki Coupling Br Br Ph3P Pd