Reductive Elimination • Migratory Insertion • - Hydrogen Elimination 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bimetallic Catalyst Catalyzed Carbonylation of Methanol to Acetic Acid
materials Article Study on Rh(I)/Ru(III) Bimetallic Catalyst Catalyzed Carbonylation of Methanol to Acetic Acid Shasha Zhang 1, Wenxin Ji 1,2,*, Ning Feng 2, Liping Lan 1, Yuanyuan Li 1,2 and Yulong Ma 1,2 1 College of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China; [email protected] (S.Z.); [email protected] (L.L.); [email protected] (Y.L.); [email protected] (Y.M.) 2 State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, Ningxia University, Yinchuan 750021, China; [email protected] * Correspondence: [email protected]; Tel.: +86-135-1957-9989; Fax: +86-951-206-2323 Received: 13 July 2020; Accepted: 3 September 2020; Published: 11 September 2020 Abstract: In this study, a Rh(I)/Ru(III) catalyst with a bimetallic space structure was designed and synthesized. The interaction between the metals of the bimetallic catalyst and the structure of the bridged dimer can effectively reduce the steric hindrance effect and help speed up the reaction rate while ensuring the stability of the catalyst. X-ray photoelectron spectroscopy (XPS) results show that rhodium accepts electrons from chlorine, thereby increasing the electron-rich nature of rhodium and improving the catalytic activity. This promotes the nucleophilic reaction of the catalyst with methyl iodide and reduces the reaction energy barrier. The methanol carbonylation performance of the Rh/Ru catalyst was evaluated, and the results show that the conversion rate of methyl acetate and the yield of acetic acid are 96.0% under certain conditions. Furthermore, during the catalysis, no precipitate is formed and the amount of water is greatly reduced. -

Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, and a FEW CASE STUDIES
Safety Moment TYLER LAB GROUP MEETING 1 Safety Moment TYLER LAB GROUP MEETING 2 Metal Hydrides: Benchtop vs. Box Hydride = :H- Hydrides are powerful Lewis bases and reducing agent ◦ Exothermically form H2 (this should scare you) ◦ Heating leads to faster reactivity ◦ H evolution leads to rapid increase in pressure2 ◦ Uncontrolled reactions easily cause runaway exotherm, class D fire, explosion, and death/unemployment LiAlH is the #1 chemical cause of fatality in chemical4 industry 3 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” LiAlH4 ◦ Insanely irritating (serious safety hazard) ◦ Extremely moisture sensitive (don’t leave out for >2 minutes) ◦ Ethereal mixtures are pyrophoric! DiBuAl-H ◦ Pyrophoric – it will explode upon exposure to oxygen NaEt3BH ◦ Pyrophoric in solution LiH and NaH ◦ Can be handled on the benchtop (not >2 minutes) ◦ Parrafin oil dispersions much safer KH ◦ Pyrophoric if not in a dispersion ◦ Handle with extreme care! † Sirius Black, Harry Potter and the Prisoner of Azkaban 4 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” CaH2 ◦ Very safe to handle on the benchtop Pt-H, Pd-H, Ni-H ◦ All very pyrophoric NaBH4 ◦ Very safe in general Other hydrides ◦ Treat as pyrophoric ◦ Transition metal hydrides vary in hydridic strength ◦ General rule of thumb: if it does hydrogenations, it is probably pyrophoric ◦ If they’re in organics of any kind, they are probably pyrophoric † Sirius Black, Harry Potter and the Prisoner of Azkaban 5 Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, -

Organometallic and Catalysis
ORGANOMETALLIC AND CATALYSIS Dr. Malay Dolai, Assistant Professor, Department of Chemistry, Prabhat Kumar College, Contai, Purba Medinipur-721404, WB, India. 1.Introduction Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkaline, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and tin, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide (metal carbonyls), cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. In 1827, Zeise's salt is the first platinum- olefin complex: K[PtCl3(C2H4)].H2O, the first invented organometallic compound. Organometallic compounds find wide use in commercial reactions, both as homogeneous catalysis and as stoichiometric reagents For instance, organolithium, organomagnesium, and organoaluminium compounds, examples of which are highly basic and highly reducing, are useful stoichiometrically, but also catalyze many polymerization reactions. Almost all processes involving carbon monoxide rely on catalysts, notable examples being described as carbonylations. The production of acetic acid from methanol and carbon monoxide is catalyzed via metal carbonyl complexes in the Monsanto process and Cativa process. Most synthetic aldehydes are produced via hydroformylation. The bulk of the synthetic alcohols, at least those larger than ethanol, are produced by hydrogenation of hydroformylation- derived aldehydes. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Transmetallation Versus Hydride Elimination
DOI: 10.1002/chem.201102678 Transmetallation Versus b-Hydride Elimination: The Role of 1,4- Benzoquinone in Chelation-Controlled Arylation Reactions with Arylboronic Acids Christian Skçld,*[a] Jonatan Kleimark,[b] Alejandro Trejos,[a] Luke R. Odell,[a] Sten O. Nilsson Lill,[b] Per-Ola Norrby,[b] and Mats Larhed[a] Abstract: The formation of an atypical, um, which is necessary to complete the complex. The association of BQ lowers saturated, diarylated, Heck/Suzuki, catalytic cycle, this electron-deficient the free-energy barrier for transmetal- domino product produced under oxida- alkene opens up a low-energy reaction lation of the s-alkyl complex to create tive Heck reaction conditions, employ- pathway from the post-insertion s-alkyl a pathway that is energetically lower ing arylboronic acids and a chelating than the oxidative Heck reaction path- vinyl ether, has been investigated by way. Furthermore, the calculations Keywords: arylation · CÀC cou- DFT calculations. The calculations showed that the reaction is made pling · density functional calcula- highlight the crucial role of 1,4-benzo- viable by BQ-mediated reductive elimi- tions · elimination · palladium · quinone (BQ) in the reaction. In addi- nation and leads to the saturated di- transmetallation tion to its role as an oxidant of palladi- arylatedACHTUNGRE product. Introduction step of the reaction, it was initially performed by using stoi- ACHTUNGRE chiometric amounts of Pd(OAc)2. More recent develop- The Mizoroki–Heck reaction[1] is a convenient and versatile ments have rendered the reaction catalytic by the re-oxida- method for the formation of carbon–carbon bonds through tion of Pd0 to PdII and today the oxidative Heck reaction is the vinylation or arylation of alkenes.[2] The reaction is per- an established and useful variant of the vinylic substitution formed by Pd0 catalysis in which the active PdII–vinyl or reaction.[4] –aryl intermediate is produced by oxidative addition of aryl One strategy to achieve regio- and stereoselectivity in the halides or pseudo-halides. -

An Investigation on the Effects of Iodide Salts in Iridium Catalyst in Methanol Carbonylation Process
Archive of SID An investigation on the effects of iodide salts in iridium catalyst in methanol carbonylation process M.T. Ashtiany, A.M. Rezaee and M.R. Jafari Nasr* Petrochemical Research & Technology Company (NPC-RT), Tehran, Iran Recieved: May 2010; Revised: July 2010; Accepted: August 2010 Abstract: In this study, iodide salts and carbonyl complexes of specific metals used as promoters in carbonylation of methanol in iridium/iodide based catalyst. The iridium catalyzed carbonylation of methanol to acetic acid is promoted by two distinct classes of promoters; simple iodide salts of zinc, cadmium, mercury, indium and gallium and carbonyl or halocarbonyl complexes of tungsten, rhenium, osmium and ruthenium. All of these promoters are led to increase the rate of methanol carbonylation. In this experimental research, it has been observed that the effect of ruthenium promoter in rate of methanol carbonylation and high reaction rate is achieved by increasing the ruthenium concentration. The experimental tests are carried out to identify the effect of different promoters. In the presence of ruthenium and iridium, it has been observed that the maximum activity is achieved between 5 to 6 weight percent of water concentration. Keywords: Methanol carbonylation, Iridium, Promoter, Iodide salt. Introduction at 250°C and 680 bar pressure [2-3]. Monsanto dis- The global annual production capacity of acetic acid covered rhodium based catalyst for carbonylation in 2004 was about 9.3 million tones. The main uses for of methanol under relatively mild conditions rather acetic acid are in production of vinyl acetate (VAM), than previous method, 250°C operating temperature solvents in the manufacturing of the terephthalic and 680 bar pressure, and commercialized its process anhydride, acetic anhydride which mainly used to at Texas City in 1970 [4-5]. -

We Can Now Define a Ligand As Any Molecule Or Ion That Has at Least
Chapter 2 General Aspects of Carbonylation Catalysis 2.1 Introduction: A chance observation by Ludwig Mond in 1884 led to an important advance in the nickel refining industry. When he found his nickel valves were being eaten away by CO, he deliberately heated Ni powder in a stream of CO to form a volatile compound, Ni(CO)4, the first known metal carbonyl. The Mond refining process was based on the fact that the carbonyl can be decomposed to give pure nickel by further heating, see Figure 2.1. Lord Kelvin was so impressed by this result that he remarked that Mond ‘gave wings to nickel’. OC CO ∆ ∆ Ni + 4CO Ni Ni + 4CO Ore OC CO Pure Figure 2.1 Mond process to refine nickel ore. The development of organometallic chemistry has over the past years gained impetus, arising from the ability of transition metals to catalyse various kinds of organic transformations1. Since 1938 when Otto Roelen discovered the oxo synthesis2,3, work in the area of homogeneous catalysis has increased greatly due in part to work done by Adkins and Krsek4, Storch et al.5, Berty and Marko6 and Natta7. 1 B. Douglas, D.H. McDaniel, J.J. Alexander, “Concepts and Models of Inorganic Chemistry”, 2nd Ed., John Wiley & Sons, New York, 1983. 2 O. Roelen, ChED Chem. Exp. Didakt., 3, 1977, 119. 3 (a) B. Cornils, W.A. Herrmann, M. Rasch, Angew. Chem., 106, 1994, 2219. (b) B. Cornils, W.A. Herrmann, M. Rasch, Angew. Chem., Int. Ed. Engl., 33, 1994, 2144. 4 H. Adkins, G. Krsek, J. -
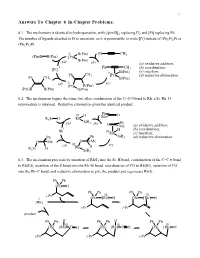
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Mechanistic Understanding of Methanol Carbonylation: Interfacing Homogeneous and Heterogeneous Catalysis Via Carbon Supported Irala
Journal of Catalysis 361 (2018) 414–422 Contents lists available at ScienceDirect Journal of Catalysis journal homepage: www.elsevier.com/locate/jcat Mechanistic understanding of methanol carbonylation: Interfacing homogeneous and heterogeneous catalysis via carbon supported IrALa Alyssa J.R. Hensley a, Jianghao Zhang a, Ilka Vinçon b, Xavier Pereira Hernandez a, Diana Tranca b, ⇑ ⇑ ⇑ Gotthard Seifert b, , Jean-Sabin McEwen a,c,d,e, , Yong Wang a,e, a The Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA 99164, United States b Theoretical Chemistry, Technische Universität Dresden, Dresden 01062, Germany c Department of Physics and Astronomy, Washington State University, Pullman, WA 99164, United States d Department of Chemistry, Washington State University, Pullman, WA 99164, United States e Institute for Integrated Catalysis, Pacific Northwest National Laboratory, Richland, WA 99352, United States article info abstract Article history: The creation of heterogeneous analogs to homogeneous catalysts is of great importance to many indus- Received 17 July 2017 trial processes. Acetic acid synthesis via the carbonylation of methanol is one such process and it relies on Revised 21 February 2018 a difficult-to-separate homogeneous Ir-based catalyst. Using a combination of density functional theory Accepted 22 February 2018 (DFT) and attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, we determine Available online 5 April 2018 the structure and mechanism for methanol carbonylation over a promising single-site IrALa/C heteroge- neous catalyst replacement. Here, the Ir center is the active site with the acetyl-Ir complex being a rate Keywords: controlling intermediate. Furthermore, the La both atomically disperses the Ir and acts as a Lewis acid Single-site heterogeneous catalyst site. -

The Emergence of Transition Metal-Mediated Hydrothiolation of Unsaturated Carbon-Carbon Bonds: a Mechanistic Outlook Ricardo Castarlenas,* Andrea Di Giuseppe, Jesús J
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital.CSIC Hydrothiolation of Unsaturated Bonds The Emergence of Transition Metal-Mediated Hydrothiolation of Unsaturated Carbon-Carbon Bonds: A Mechanistic Outlook Ricardo Castarlenas,* Andrea Di Giuseppe, Jesús J. Pérez-Torrente, Luis A. Oro* The hydrothiolation of unsaturated carbon-carbon bonds is a practical and atom-economical approach for the incorporation of sulfur into organic frameworks. In recent years, we have witnessed the development of a range of transition metal-based catalytic systems for the control of the regio- and stereoselectivity. This minireview aims to highlight the mechanistic background behind this transformation in order to help for the design of more specific and active organometallic hydrothiolation catalysts. 1. Introduction in-depth understanding of the mechanistic issues, and indeed, it could be useful to other diverse hydroelementation transformations The development of efficient synthetic methods for the such as hydroalkoxylation, hydrophosphination, hydroamination, or incorporation of sulfur into organic frameworks is nowadays an hydroacylation, among others. Our aim in this concise survey is to important task due to the practical applications of this type of analyze the different mechanistic pathways rather than to present a compounds as pharmaceuticals, functional materials, or synthetic comprehensive overview of a research area that has already been reagents.[1-4] In this context, one of the simplest and atom- covered in several recent reviews.[1,5] The sulfa-michael additions economical approach is the direct addition of sulfur and hydrogen mediated by organocatalysts remains beyond the scope of this atoms from thiols to unsaturated carbon-carbon bonds, commonly minireview. -

Insertion Reactions • Oxidative Addition and Substitution Allow Us to Assemble 1E and 2E Ligands on the Metal, Respectively
Insertion Reactions • Oxidative addition and substitution allow us to assemble 1e and 2e ligands on the metal, respectively. • With insertion, and its reverse reaction, elimination, we can now combine and transform these ligands within the coordination sphere, and ultimately expel these transformed ligands to form free organic compounds. • There are two main types of insertion 1) 1,1 insertion in which the metal and the X ligand end up bound to the same (,)(1,1) atom 2) 1,2 insertion in which the metal and the X ligand end up bound to adjacent (1,2) atoms of an L‐type ligand. • The type of insertion observed in any given case depends on the nature of the 2e inserting ligand. • For example: CO gives only 1,1 insertion ethylene gives only 1,2 insertion, in which the M and the X end up on adjacent atoms of what was the 2e X‐type ligand. In general, η1 ligands tend to give 1,1 insertion and η2 ligands give 1,2 insertion • SO2 is the only common ligan d tha t can give bthboth types of itiinsertion; as a ligan d, SO2 can be η1 (S) or η2 (S, O). • In principle, insertion reactions are reversible, but just as we saw for oxidative addition and reductive elimination previously, for many ligands only one of the two possible directions is observed in practice, probably because this direction is strongly favored thermodynamically. •A2e vacant site is generated by 1,1 and 1,2 insertion reactions. • Thissite can be occupidied byanextlternal 2e ligan d and the itiinsertion prodtduct tdtrapped. -

Solventless Migratory-Insertion Reactions of Substituted Cyclopentadienyl Iron Complexes Induced by Electron Donor Ligands
Bull. Chem. Soc. Ethiop. 2009 , 23(3), 399-407. ISSN 1011-3924 Printed in Ethiopia 2009 Chemical Society of Ethiopia SOLVENTLESS MIGRATORY-INSERTION REACTIONS OF SUBSTITUTED CYCLOPENTADIENYL IRON COMPLEXES INDUCED BY ELECTRON DONOR LIGANDS Apollinaire Munyaneza 1, Olalere G. Adeyemi 1, 2 and Neil J. Coville 1* 1Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg, 2050, South Africa 2Department of Chemical Sciences, College of Natural Sciences, Redeemer’s University, Km 46, Lagos/Ibadan Expressway, Redemption City, Nigeria (Received February 24, 2009; revised July 2, 2009) ABSTRABSTRACT.ACT. Reaction between solid (C 5H5)Fe(CO) 2CH 3 and a range of solid phosphine ligands, L (L = PPh 3, P( m-CH 3C6H4)3, P( p-CH 3C6H4)3, P( p-FC 6H4)3, P( p-ClC 6H4)3, PCy 3) occurred in the absence of solvent in the melt phase to give the migratory-insertion products, (C 5H5)Fe(CO)(COCH 3)(L). The reaction was more rapid with small electron withdrawing ligands. Insertion reaction between (RC 5H4)Fe(CO) 2R’ (R = H, CH 3; R’ = CH 3, CH 2Ph) and gaseous ligands such as SO 2 and CO were also studied. The insertion of SO 2 occurred readily for all the substrates investigated, but CO insertion did not occur (< 1 %) using the solventless reaction condition. KEY WORDS: Melt phase, Solventless chemistry, Cyclopentadiene, Iron complexes, Migratory-insertion INTRODUCTION An insertion reaction occurs when an incoming ligand is incorporated into an existing complex without releasing any of the previously coordinated groups [1]. Generally, small molecules such as CO, SO 2, carbenes, etc.