Computational Aspects of Hydroformylation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2. Catalysis Involving CO
2. Catalysis Involving CO (Source: Collman / Hegedus + Chiusoli / Maitlis + original papers mentioned below) !1 General Reactivity of CO-Complexes 1 is the resonance representing the pure "-donation of CO to the metal. 3 is contributing the most when the # back donation from the metal to CO is weak. The carbon is more electrophilic here. 2 is the extreme structure that evidences the # back donation of the metal to the #* of CO. !2 – Synthesis Gas and Water Gas Shift Reaction CO / H2 as feedstock. – Hydrocarbonylation (or Hydroformylation) of Olefins / Oxo Reaction Synthesis of aldehydes and alcohols from alkenes with cobalt and rhodium catalysts. – Carbonylation of Alcohols: Monsanto’s Acetic Acid Process Preparation of acetic acid from methanol and CO. !3 Synthesis Gas (Syn Gas, CO / H2) as Feedstock Steam over coal: C + H2O → CO + H2 0 0 (!ΔH 298 K = 131 kJ/mol; !ΔG1073 K= –12 kJ/mol) Steam reforming of methane: CH4 + H2O → CO + 3 H2 0 0 (!ΔH 298 K = 206 kJ/mol; !ΔG1073 K= –24 kJ/mol) Coupled with partial oxidation to give an endothermic overall reaction: H 0 2 C + H2O + O2 → CO + CO2 + H2 (Δ 298 K = –285 kJ/mol) 1 H 0 CH4 + 2 O2 → CO + 2 H2 (Δ 298 K = –36 kJ/mol) !4 Water-Gas-Shift Reaction (WGSR) Allows adjust the CO : H2 ratio by converting CO to H2: !!⇀ CO + H2O ↽!! CO2 + H2 Drawback: CO2 as byproduct. Catalysts: Heterogeneous Cr2O3 (T = 350°C) Cu-Zn-oxide (T = 200 – 300°C) Fe3O4 Homogeneous Carbonyl complexes: [FeH(CO)4]–, [RhI2(CO)2]–, [RuCl(bipy)2(CO)]+ !5 Homogeneously Catalyzed WGSR Principle: O CO OH– – CO2 M M CO M C M H – OH – + H O – ! M H 2 M + OH + H2 !6 Catalytic WGSR ! !7 Hydroformylation or Oxo Synthesis Synthesis of aldehydes and alcohols from alkenes. -

Reactions of Alkenes and Alkynes with Formaldehyde Catalyzed by Rhodium Systems Containing Phosphine Ligands
J. Mex. Chem. Soc. 2017, 61(2), 120-127 Article © 2017, Sociedad Química de México ISSN 1870-249X Reactions of alkenes and alkynes with formaldehyde catalyzed by rhodium systems containing phosphine ligands Merlín Rosales,1* Beatriz González,1 Jessely Molina,1 Homero Pérez,1 María Modroño-Alonso2 and Pablo J. Baricelli2 1 Universidad del Zulia (L.U.Z.), Facultad Experimental de Ciencias. Departamento de Química, Laboratorio de Química Inorgánica (LQI). Maracaibo (Venezuela). 2 Universidad de Carabobo, Facultad de Ingeniería, Centro de Investigaciones Químicas, Valencia (Venezuela). * Corresponding author: Tel +584143602104 FAX +582614127701 Ciudad Universitaria. Módulo 2. Maracaibo. Venezuela e-mail adress: merlin2002 @cantv.net; [email protected] (M. Rosales) Received October 18th, 2016; Accepted March 8th, 2017. Abstract. The reaction of alkenes (allyl alcohol, styrene and C6 Resumen. La reacción de alquenos (alcohol alílico, estireno y alquenos alkenes) with formaldehyde was efficiently performed by using Rh C6) con formaldehido se realizó eficientemente usando precatalizado- precatalysts formed in situ by the addition of triphenylphosphine res de Rh formados in situ por adición de trifenilfosfina (PPh3), (PPh3), 1,2-bis(diphenylphosphino)ethane (dppe) or 1,1,1-tris(diphen- 1,2-bis(difenilfosfino)etano (dffe) o 1,1,1-tris(difenilfosfinometil) etano ylphosphinomethyl)ethane (triphos) to the complex Rh(acac)(CO)2 at (trifos) al complejo Rh(acac)(CO)2 a 130ºC en 1,4-dioxano, produ- 130ºC in 1,4-dioxane, yielding their corresponding aldehydes; the best ciendo los correspondientes aldehídos. El mejor sistema catalítico fue 2 catalytic system was Rh(acac)(CO)2/2dppe, which generates the cat- Rh(acac)(CO)2/2dffe, el cual genera el complejo catiónico [Rh(k -P,P- 2 + + ionic complex [Rh(k -P,P-dppe)2] . -

Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, and a FEW CASE STUDIES
Safety Moment TYLER LAB GROUP MEETING 1 Safety Moment TYLER LAB GROUP MEETING 2 Metal Hydrides: Benchtop vs. Box Hydride = :H- Hydrides are powerful Lewis bases and reducing agent ◦ Exothermically form H2 (this should scare you) ◦ Heating leads to faster reactivity ◦ H evolution leads to rapid increase in pressure2 ◦ Uncontrolled reactions easily cause runaway exotherm, class D fire, explosion, and death/unemployment LiAlH is the #1 chemical cause of fatality in chemical4 industry 3 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” LiAlH4 ◦ Insanely irritating (serious safety hazard) ◦ Extremely moisture sensitive (don’t leave out for >2 minutes) ◦ Ethereal mixtures are pyrophoric! DiBuAl-H ◦ Pyrophoric – it will explode upon exposure to oxygen NaEt3BH ◦ Pyrophoric in solution LiH and NaH ◦ Can be handled on the benchtop (not >2 minutes) ◦ Parrafin oil dispersions much safer KH ◦ Pyrophoric if not in a dispersion ◦ Handle with extreme care! † Sirius Black, Harry Potter and the Prisoner of Azkaban 4 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” CaH2 ◦ Very safe to handle on the benchtop Pt-H, Pd-H, Ni-H ◦ All very pyrophoric NaBH4 ◦ Very safe in general Other hydrides ◦ Treat as pyrophoric ◦ Transition metal hydrides vary in hydridic strength ◦ General rule of thumb: if it does hydrogenations, it is probably pyrophoric ◦ If they’re in organics of any kind, they are probably pyrophoric † Sirius Black, Harry Potter and the Prisoner of Azkaban 5 Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, -

Hydroformylation of Formaldehyde with Rhodium Catalyst
Patentamt O JEuropâischesEuropean Patent Office © Publication number: 002 908 Office européen des brevets Bl @ EUROPEAN PATENT SPECIFICATION (46) Date of publication of patent spécification: 12.05.82 © Int. Cl.3: C 07 C 47/19, 8 01 J 31/°2' C 07 C 45/50 © Application number: 78300820.4 @ Date of filing: 1 4.1 2.78 © Hydroformylation of formaldehyde with rhodium cataiyst. © Priority: 16.12.77 US 861474 @ Proprietor: MONSANTO COMPANY Patent Department 800 North Lindbergh Boulevard © Date of publication of application: St. Louis, Missouri 631 66 (US) 11.07.79 Bulletin 79/14 @ Inventor: Spencer, Alwyn © Publication of the grant of the patent: Johnson Matthey Research Centre Blount's Court 1 2.05.82 Bulletin 82/1 9 Jonning Common Reading R64-9NH (GB) @ Designated Contracting States: © Representative: Lunt, John Cooper etal, BECHDEFRGBLUNLSE Monsanto House 1 0-1 8 Victoria Street London, SW1H ONQ(GB) "© References cited: DE- A- 3 940 432 DE - A - 2 741 589 GB - A - 1 335 531 US - A - 3 940 432 Chemical Abstracts vol. 87, no. 19. 1977, Columbus, Ohio, USA T. MIZOROKI "Syntheses of oxygen-containing C2-compounds using carbon monoxide" page 541, column 1, abstract no. 151 634g Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition shall be filed in a written reasoned statement. It shall not be deemed to have been filed until the opposition fee has been paid. (Art. -

Hydroformylation of 1-Hexene Over Rh/Nano-Oxide Catalysts
Catalysts 2013, 3, 324-337; doi:10.3390/catal3010324 OPEN ACCESS catalysts ISSN 2073-4344 www.mdpi.com/journal/catalysts Article Hydroformylation of 1-Hexene over Rh/Nano-Oxide Catalysts Maija-Liisa Kontkanen 1, Matti Tuikka 1,2, Niko M. Kinnunen 1, Sari Suvanto 1 and Matti Haukka 1,2,* 1 Department of Chemistry, University of Eastern Finland, P.O. Box 111, FI-80101 Joensuu, Finland; E-Mails: [email protected] (M.-L.K.); [email protected] (M.T.); [email protected] (N.M.K.); [email protected] (S.S.) 2 Department of Chemistry, University of Jyväskylä, P.O. Box 35, University of Jyväskylä, Finland * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +358-40-805-4666; Fax: +358-4-260-2501. Received: 26 January 2013; in revised form: 25 February 2013 / Accepted: 6 March 2013 / Published: 21 March 2013 Abstract: The effect of nanostructured supports on the activity of Rh catalysts was studied by comparing the catalytic performance of nano- and bulk-oxide supported Rh/ZnO, Rh/SiO2 and Rh/TiO2 systems in 1-hexene hydroformylation. The highest activity with 100% total conversion and 96% yield of aldehydes was obtained with the Rh/nano-ZnO catalyst. The Rh/nano-ZnO catalyst was found to be more stable and active than the corresponding rhodium catalyst supported on bulk ZnO. The favorable morphology of Rh/nano-ZnO particles led to an increased metal content and an increased number of weak acid sites compared to the bulk ZnO supported catalysts. Both these factors favored the improved catalytic performance. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Hydroformylation of C3 and C8 Olefins in Hydrocarbon Gas-Expanded Solvents
HYDROFORMYLATION OF C3 AND C8 OLEFINS IN HYDROCARBON GAS-EXPANDED SOLVENTS By Dupeng Liu Submitted to the graduate degree program in Chemical & Petroleum Engineering and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Chairperson: Bala Subramaniam Raghunath V. Chaudhari Jon Tunge Aaron Scurto Kevin Leonard Date Defended: April 30th, 2018 The dissertation committee for Dupeng Liu certifies that this is the approved version of the following dissertation: HYDROFORMYLATION OF C3 AND C8 OLEFINS IN HYDROCARBON GAS-EXPANDED SOLVENTS Chairperson: Bala Subramaniam Date Approved: May 8th, 2018 ii Abstract Hydroformylation involves the addition of syngas to the double bond of an alkene yielding aldehydes used to produce basic building blocks for myriad consumer goods. Resource-efficient technologies that conserve feedstock and energy continue to be of interest to industry. It has been shown previously by our group that the use of CO2-expanded liquids significantly enhances the Rh-catalyzed 1-octene hydroformylation rate and selectivity. This dissertation extends this concept by employing light alkanes such as propane and n-butane as compressed solvents in propylene and 1-octene hydroformylation, respectively. Phase behavior measurements demonstrate that at identical H2 or CO fugacities in the vapor phase, the H2 and CO solubilities in either propane- or propylene-expanded solvents (PXLs) are greater than those in the corresponding neat solvents by as high as 76% at 70 °C and 1.5 MPa. The H2/CO ratio in PXLs is enhanced by simply increasing the propane partial pressure. In contrast, the H2/CO in the neat solvent at a given temperature is constant at all syngas partial pressures. -

Transmetallation Versus Hydride Elimination
DOI: 10.1002/chem.201102678 Transmetallation Versus b-Hydride Elimination: The Role of 1,4- Benzoquinone in Chelation-Controlled Arylation Reactions with Arylboronic Acids Christian Skçld,*[a] Jonatan Kleimark,[b] Alejandro Trejos,[a] Luke R. Odell,[a] Sten O. Nilsson Lill,[b] Per-Ola Norrby,[b] and Mats Larhed[a] Abstract: The formation of an atypical, um, which is necessary to complete the complex. The association of BQ lowers saturated, diarylated, Heck/Suzuki, catalytic cycle, this electron-deficient the free-energy barrier for transmetal- domino product produced under oxida- alkene opens up a low-energy reaction lation of the s-alkyl complex to create tive Heck reaction conditions, employ- pathway from the post-insertion s-alkyl a pathway that is energetically lower ing arylboronic acids and a chelating than the oxidative Heck reaction path- vinyl ether, has been investigated by way. Furthermore, the calculations Keywords: arylation · CÀC cou- DFT calculations. The calculations showed that the reaction is made pling · density functional calcula- highlight the crucial role of 1,4-benzo- viable by BQ-mediated reductive elimi- tions · elimination · palladium · quinone (BQ) in the reaction. In addi- nation and leads to the saturated di- transmetallation tion to its role as an oxidant of palladi- arylatedACHTUNGRE product. Introduction step of the reaction, it was initially performed by using stoi- ACHTUNGRE chiometric amounts of Pd(OAc)2. More recent develop- The Mizoroki–Heck reaction[1] is a convenient and versatile ments have rendered the reaction catalytic by the re-oxida- method for the formation of carbon–carbon bonds through tion of Pd0 to PdII and today the oxidative Heck reaction is the vinylation or arylation of alkenes.[2] The reaction is per- an established and useful variant of the vinylic substitution formed by Pd0 catalysis in which the active PdII–vinyl or reaction.[4] –aryl intermediate is produced by oxidative addition of aryl One strategy to achieve regio- and stereoselectivity in the halides or pseudo-halides. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0032845 A1 Han Et Al
US 20030032845A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0032845 A1 Han et al. (43) Pub. Date: Feb. 13, 2003 (54) HYDROFORMYLATION OF ETHYLENE Related U.S. Application Data OXIDE (63) Continuation-in-part of application No. 09/924,822, (76) Inventors: Yuan-Zhang Han, West Chester, PA filed on Aug. 8, 2001, now abandoned. (US); Krishnan Viswanathan, Houston, TX (US) Publication Classification Correspondence Address: (51) Int. Cl. ................................................ C07C 29/15 LYONDELL CHEMICAL COMPANY (52) U.S. Cl. ............................................ 568/862; 568/867 3801 WEST CHESTER PIKE NEWTOWN SQUARE, PA 19073 (US) (57) ABSTRACT (21) Appl. No.: 10/038,975 1,3-propanediol is formed from ethylene oxide containing formaldehyde and acetaldehyde impurities by hydroformy (22) Filed: Jan. 4, 2002 lation and hydrogenation. Patent Application Publication Feb. 13, 2003 US 2003/0032845 A1 N y so US 2003/0032845 A1 Feb. 13, 2003 HYDROFORMYLATION OF ETHYLENE OXDE wise, the procedures described in recently filed applications Ser. No. 09/882,346, 09/882,347 and 09/882,641 filed Jun. RELATED APPLICATION 15, 2001 can be employed and are incorporated herein by 0001. This application is a continuation-in-part of appli reference. cation Ser. No. 09/924,822 filed Aug. 8, 2001. 0010 Generally speaking, ethylene oxide which has been employed as feed in prior procedures is a commercial grade BACKGROUND OF THE INVENTION of ethylene oxide from which a predominance of the impu rities coproducted during ethylene oxide formation have 0002) 1. Field of the Invention been removed. Generally, the aldehyde Specifications for 0003. This invention relates to the catalytic hydroformy such commercial ethylene oxide are a maximum of 30-50 lation of ethylene oxide and especially to a process wherein ppm by weight of aldehyde expressed as acetaldehyde. -

And Regioselective Hydroformylation of Alkenes with CO2/H2 Over a Bifunctional Catalyst
Chemo- and Regioselective Hydroformylation of Alkenes with CO2/H2 over a Bifunctional Catalyst Kai HUA Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210 (P. R. China). Fang Liu Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210 (P. R. China). Yin Wei Low-Carbon Conversion Science and Engineering Long Shao Shanghai Advanced Research Institute, Chinese Academy of Sciences Chao Deng ShanghaiTech University Ming Zhang Shanghai Advanced Research Institute, Chinese Academy of Sciences Lin Xia Shanghai Advanced Research Institute, Chinese Academy of Sciences Liangshu Zhong Shanghai Advanced Research Institute https://orcid.org/0000-0002-4167-8630 Hui Wang ( [email protected] ) Shanghai Advanced Research Institute, Chinese Academy of Sciences Yuhan Sun Shanghai Advanced Research Institute, Chinese Academy of Sciences Article Keywords: CO2, H2, chemo- and regioselective hydroformylation of alkenes, bifunctional catalyst Posted Date: October 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-85641/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Green Chemistry on January 1st, 2021. See the published version at https://doi.org/10.1039/D0GC03913F. Chemo- and Regioselective Hydroformylation of Alkenes with CO2/H2 over a Bifunctional Catalyst Kaimin Hua1,2, Xiaofang Liu1,* Baiyin Wei1,3, Zilong Shao1, Yuchao Deng1,3, Jianming Zhang1, Lin Xia1, Liangshu Zhong1,3, Hui Wang1,* Yuhan Sun1,3,4* Abstract: Combining CO2 and H2 to prepare building blocks for high-value-added products is an attractive yet challenging approach. A general and selective rhodium- catalyzed hydroformylation of alkenes using CO2/H2 as a syngas surrogate is described here. -
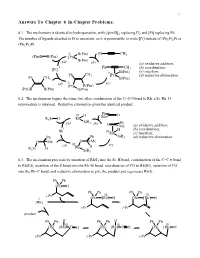
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Preparation and Characterization of Rh/Mgsnts Catalyst for Hydroformylation of Vinyl Acetate: the Rh0 Was Obtained by Calcination
Article Preparation and Characterization of Rh/MgSNTs Catalyst for Hydroformylation of Vinyl Acetate: The Rh0 was Obtained by Calcination Penghe Su 1, Ya Chen 1, Xiaotong Liu 1, Hongyuan Chuai 1, Hongchi Liu 1, Baolin Zhu 1,2, Shoumin Zhang 1,2 and Weiping Huang 1,2,3,* 1 College of Chemistry, Nankai University, Tianjin 300071, China; [email protected] (P.S.); [email protected] (Y.C.); [email protected] (X.L.); [email protected] (H.C.); [email protected] (H.L.); [email protected] (B.Z.); [email protected] (S.Z.) 2 The Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China 3 Collaborative Innovation Center of Chemical Science and Engineering, Tianjin 300071, China * Correspondence: [email protected]; Tel.: +86-138-2009-6974 Received: 01 February 2019; Accepted: 18 February 2019; Published: 26 February 2019 Abstract: A simple and practical Rh-catalyzed hydroformylation of vinyl acetate has been synthesized via impregnation-calcination method using silicate nanotubes (MgSNTs) as the supporter. The Rh0 (zero valent state of rhodium) was obtained by calcination. The influence of calcination temperature on catalytic performance of the catalysts was investigated in detail. The catalysts were characterized in detail by X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectrometer (XPS), atomic emission spectrometer (ICP), and Brunauer–Emmett–Teller (BET) surface-area analyzers. The Rh/MgSNTs(a2) catalyst shows excellent catalytic activity, selectivity and superior cyclicity. The catalyst could be easily recovered by phase separation and was used up to four times. Keywords: silicate nanotubes; Rh; hydroformylation; vinyl acetate 1.