Exploring the Mechanism of the Pd-Catalyzed Spirocyclization Reaction: a Combined DFT and Cite This: Chem
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Insertion Reactions • Oxidative Addition and Substitution Allow Us to Assemble 1E and 2E Ligands on the Metal, Respectively
Insertion Reactions • Oxidative addition and substitution allow us to assemble 1e and 2e ligands on the metal, respectively. • With insertion, and its reverse reaction, elimination, we can now combine and transform these ligands within the coordination sphere, and ultimately expel these transformed ligands to form free organic compounds. • There are two main types of insertion 1) 1,1 insertion in which the metal and the X ligand end up bound to the same (,)(1,1) atom 2) 1,2 insertion in which the metal and the X ligand end up bound to adjacent (1,2) atoms of an L‐type ligand. 1 • The type of insertion observed in any given case depends on the nature of the 2e inserting ligand. • For example: CO gives only 1,1 insertion ethylene gives only 1,2 insertion, in which the M and the X end up on adjacent atoms of what was the 2e X‐type ligand. In general, η1 ligands tend to give 1,1 insertion and η2 ligands give 1,2 insertion • SO2 is the only common ligan d tha t can give bthboth types of itiinsertion; as a ligan d, SO2 can be η1 (S) or η2 (S, O). • In principle, insertion reactions are reversible, but just as we saw for oxidative addition and reductive elimination previously, for many ligands only one of the two possible directions is observed in practice, probably because this direction is strongly favored thermodynamically. 2 •A2e vacant site is generated by 1,1 and 1,2 insertion reactions. • Thissite can be occupidied byanextlternal 2e ligan d and the itiinsertion prodtduct tdtrapped. -

Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, and a FEW CASE STUDIES
Safety Moment TYLER LAB GROUP MEETING 1 Safety Moment TYLER LAB GROUP MEETING 2 Metal Hydrides: Benchtop vs. Box Hydride = :H- Hydrides are powerful Lewis bases and reducing agent ◦ Exothermically form H2 (this should scare you) ◦ Heating leads to faster reactivity ◦ H evolution leads to rapid increase in pressure2 ◦ Uncontrolled reactions easily cause runaway exotherm, class D fire, explosion, and death/unemployment LiAlH is the #1 chemical cause of fatality in chemical4 industry 3 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” LiAlH4 ◦ Insanely irritating (serious safety hazard) ◦ Extremely moisture sensitive (don’t leave out for >2 minutes) ◦ Ethereal mixtures are pyrophoric! DiBuAl-H ◦ Pyrophoric – it will explode upon exposure to oxygen NaEt3BH ◦ Pyrophoric in solution LiH and NaH ◦ Can be handled on the benchtop (not >2 minutes) ◦ Parrafin oil dispersions much safer KH ◦ Pyrophoric if not in a dispersion ◦ Handle with extreme care! † Sirius Black, Harry Potter and the Prisoner of Azkaban 4 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” CaH2 ◦ Very safe to handle on the benchtop Pt-H, Pd-H, Ni-H ◦ All very pyrophoric NaBH4 ◦ Very safe in general Other hydrides ◦ Treat as pyrophoric ◦ Transition metal hydrides vary in hydridic strength ◦ General rule of thumb: if it does hydrogenations, it is probably pyrophoric ◦ If they’re in organics of any kind, they are probably pyrophoric † Sirius Black, Harry Potter and the Prisoner of Azkaban 5 Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, -

Insertion Reactions of Isocyanides Into the Metal-C(Sp3) Bonds of Ylide Complexes
Journal of Organometallic Chemistry 894 (2019) 61e66 Contents lists available at ScienceDirect Journal of Organometallic Chemistry journal homepage: www.elsevier.com/locate/jorganchem Insertion reactions of isocyanides into the Metal-C(sp3) bonds of ylide complexes ** * María-Teresa Chicote a, , Isabel Saura-Llamas a, , María-Francisca García-Yuste a, Delia Bautista b, Jose Vicente a a Grupo de Química Organometalica, Departamento de Química Inorganica, Facultad de Química, Universidad de Murcia, Spain b ACTI, Universidad de Murcia, E-30100, Murcia, Spain article info abstract Article history: The synthesis of the previously reported complexes [Pd{(C,CeCHCO2R)2PPh2}(m-Cl)]2 (R ¼ Me (1), Et (2)) Received 10 January 2019 has been considerably improved by reacting the phosphonium salts [Ph2P(CH2CO2R)2]Cl with Pd(OAc)2. Received in revised form t Complexes 1 and 2 react with isocyanides R'NC (R’ ¼ C6H4Me2-2,6 (Xy), C6H4I-2 and Bu) to give com- 29 April 2019 plexes of the type [Pd{C,C-{C(CO R) ¼ C(NHR0)}(CHCO R)}PPh }Cl(CNR0)], resulting from the insertion of Accepted 5 May 2019 2 2 2 the unsaturated molecule in one of their PdeC bonds and the hydrogen transfer from the methine CH to Available online 11 May 2019 the iminobenzoyl nitrogen. These are the first insertion reactions observed in the PdeC bond of a P-ylide complex. The crystal structure of the complex [Pd{C,C-{C(CO Me)¼C(NHXy)}(CHCO Me)}PPh }Cl(CNXy)] Keywords: 2 2 2 Palladacycles (3) is reported. © Insertion reactions 2019 Elsevier B.V. All rights reserved. -

Recent Advances on Mechanistic Studies on C–H Activation
Open Chem., 2018; 16: 1001–1058 Review Article Open Access Daniel Gallego*, Edwin A. Baquero Recent Advances on Mechanistic Studies on C–H Activation Catalyzed by Base Metals https:// doi.org/10.1515/chem-2018-0102 received March 26, 2018; accepted June 3, 2018. 1Introduction Abstract: During the last ten years, base metals have Application in organic synthesis of transition metal- become very attractive to the organometallic and catalytic catalyzed cross coupling reactions has been positioned community on activation of C-H bonds for their catalytic as one of the most important breakthroughs during the functionalization. In contrast to the statement that new millennia. The seminal works based on Pd–catalysts base metals differ on their mode of action most of the in the 70’s by Heck, Noyori and Suzuki set a new frontier manuscripts mistakenly rely on well-studied mechanisms between homogeneous catalysis and synthetic organic for precious metals while proposing plausible chemistry [1-5]. Late transition metals, mostly the precious mechanisms. Consequently, few literature examples metals, stand as the most versatile catalytic systems for a are found where a thorough mechanistic investigation variety of functionalization reactions demonstrating their have been conducted with strong support either by robustness in several applications in organic synthesis [6- theoretical calculations or experimentation. Therefore, 12]. Owing to the common interest in the catalysts mode we consider of highly scientific interest reviewing the of action by many research groups, nowadays we have a last advances on mechanistic studies on Fe, Co and Mn wide understanding of the mechanistic aspects of precious on C-H functionalization in order to get a deep insight on metal-catalyzed reactions. -

Abstract(Doctor)
別紙4-1(課程博士(英文)) Date of Submission(month day,year): March 27, 2020 Department Applied Chemistry and Life Student ID Number D 169403 SEIJI IWASA Science Supervisors KAZUTAKA SHIBATOMI Applicant’s name PHAN THI THANH NGA Abstract(Doctor) Catalytic Intramolecular Carbene Transfer Reactions into σ and π Bonds Title of Thesis (σ 及びπ結合への触媒的分子内カルベン移動反応) Approx. 800 words A carbene known as an most active intermediate is complexed with a transition metal, which afford the corresponding metal-carbene complex and catalytically insert into σ and π bonds of organic compound. Even though there are many reports on the carbene transfer process to develop a new approach for the synthesis of medicine and other bioactive compounds, the regio-, stereo- and chemoselective approaches are still limited and remained as a main subject in the filed of synthetic organic chemistry. For these background, I developed an efficient catalytic intramolecular carbene transfer reactions by using originally developed ruthenium catalyst into σ and π bonds and successfully applied for the synthesis of γ-lactam ring fused aromatics (oxindoles), γ-lactone ring fused cyclopropanes, and γ-lactam ring fused seven member rings via Buchner reaction. Although ruthenium complex is a newcomer in the field of catalytic carbene transfer reaction, it has emerged as a useful transition metal for the carbenoid chemistry of diazo compounds, besides copper and rhodium. And recently, we have developed a Ru(II)-Pheox complex, which is efficient for carbene transfer reactions, in particular, asymmetric cyclopropanation, N-H insertion, C-H insertion and Si-H insertion reactions. Therefore, driven by my interests in the catalytic asymmetric carbene transfer reaction and the efficiency displayed by the Ru(II)-Pheox catalyst, I started to explore the asymmetric cyclopropanation, C-H insertion, Buchner reactions of various diazo compounds, which are potentially building blocks and expectant to be applied in pharmaceutical and medicinal fields . -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Carbene Rearrangements: Intramolecular Interaction of a Triple Bond with a Carbene Center
An Abstract OF THE THESIS OF Jose C. Danino for the degree of Doctor of Philosophy in Chemistry presented on _Dcc, Title: Carbene RearrangementE) Intramolecular Interaction of a Triple Bond with aCarbene Center Redacted for Privacy Abstract approved: Dr. Vetere. Freeman The tosylhydrazones of2-heptanone, 4,4-dimethy1-2- heptanone, 6-heptyn-2-one and 4,4-dimethy1-6-heptyn-2- one were synthesizedand decomposed under a varietyof reaction conditions:' drylithium and sodium salt pyrolyses, sodium methoxide thermolysesin diglyme and photolyses of the lithium salt intetrahydrofuran. The saturated ana- logues 2-heptanone tosylhydrazoneand its 4,4-dimethyl isomer afforded the alkenesarising from 6-hydrogeninser- product distribution in the tion. It was determined that differ- dry salt pyrolyses of2-heptanone tosylhydrazone was ent for the lithiumand the sodium salts. However, the product distribution of thedry sodium salt was verysimilar diglyme to product distributionobtained on thermolysis in explained by a with sodium methoxide. This difference was reaction of lithium bromide(present as an impurity inall compound to the lithium salts)with the intermediate diazo afford an organolithiumintermediate that behaves in a some- what different fashionthan the free carbene.The unsaturated analogues were found to produce a cyclic product in addition to the expected acyclic alkenes arising from 3-hydrogen insertion. By comparison of the acyclic alkene distri- bution obtained in the saturated analogues with those in the unsaturated analogues, it was concluded that at leastsome cyclization was occurring via addition of the diazo moiety to the triple bond. It was determined that the organo- lithium intermediateresulting from lithium bromide cat- alyzed decomposition of the diazo compound was incapable of cyclization. -

Transmetallation Versus Hydride Elimination
DOI: 10.1002/chem.201102678 Transmetallation Versus b-Hydride Elimination: The Role of 1,4- Benzoquinone in Chelation-Controlled Arylation Reactions with Arylboronic Acids Christian Skçld,*[a] Jonatan Kleimark,[b] Alejandro Trejos,[a] Luke R. Odell,[a] Sten O. Nilsson Lill,[b] Per-Ola Norrby,[b] and Mats Larhed[a] Abstract: The formation of an atypical, um, which is necessary to complete the complex. The association of BQ lowers saturated, diarylated, Heck/Suzuki, catalytic cycle, this electron-deficient the free-energy barrier for transmetal- domino product produced under oxida- alkene opens up a low-energy reaction lation of the s-alkyl complex to create tive Heck reaction conditions, employ- pathway from the post-insertion s-alkyl a pathway that is energetically lower ing arylboronic acids and a chelating than the oxidative Heck reaction path- vinyl ether, has been investigated by way. Furthermore, the calculations Keywords: arylation · CÀC cou- DFT calculations. The calculations showed that the reaction is made pling · density functional calcula- highlight the crucial role of 1,4-benzo- viable by BQ-mediated reductive elimi- tions · elimination · palladium · quinone (BQ) in the reaction. In addi- nation and leads to the saturated di- transmetallation tion to its role as an oxidant of palladi- arylatedACHTUNGRE product. Introduction step of the reaction, it was initially performed by using stoi- ACHTUNGRE chiometric amounts of Pd(OAc)2. More recent develop- The Mizoroki–Heck reaction[1] is a convenient and versatile ments have rendered the reaction catalytic by the re-oxida- method for the formation of carbon–carbon bonds through tion of Pd0 to PdII and today the oxidative Heck reaction is the vinylation or arylation of alkenes.[2] The reaction is per- an established and useful variant of the vinylic substitution formed by Pd0 catalysis in which the active PdII–vinyl or reaction.[4] –aryl intermediate is produced by oxidative addition of aryl One strategy to achieve regio- and stereoselectivity in the halides or pseudo-halides. -

C)Ffkx.Ali%& Copy
. ~ Submitted to the Encyclopedia of Catalysis; http: //www.enccat.com/ July 2000 4 * BNL-67609 Isotope Methods in Homogeneous Catalysis R. Morris Bullock and Bruce R. Bender Chemistry Department, Brookhaven National Laboratory, Upton, New York 11973-5000 and Department of Chemistry, The Scripps Research Institute, La Jolla, CA 92110 The use of isotope labels has had a Iimdamentally important role in the determination of mechanisms of homogeneously catalyzed reactions. Mechanistic data is valuable since it can assist in the design and rational improvement of homogeneous catalysts. There are several ways to use isotopes in mechanistic chemistry. Isotopes can be introduced into controlled experiments and “followed” where they go or don’t go; in this way, Libby, Calvin, Taube and others used isotopes to elucidate mechanistic pathways for very different, yet important chemistries. Another important isotope method is the study of kinetic isotope effects (KIEs) and equilibrium isotope effect (EIEs). Here the mere observation of where a label winds up is no longer enough – what matters is how much slower (or faster) a labeled molecule reacts than the unlabeled material. The most carefid studies essentially involve the measurement of isotope fractionation between a reference ground state and the transition state. Thus kinetic isotope effects provide unique data unavailable from other methods, since information about the transition state of a reaction is obtained. Because getting an experimental glimpse of transition states is really tantamount to understanding catalysis, kinetic isotope effects are very powerfid. Direct substitution of D for H (e.g., D2 vs. H2, or D+vs. H’_)and measurement of the rate of a catalytic reaction provides a determination of the kinetic deuterium isotope effect on the overall reaction. -
Answers to Chapter 6 In-Chapter Problems
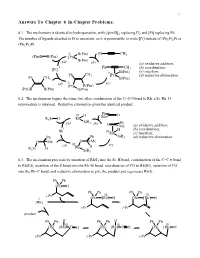
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Towards the Synthesis of Isocoronene
Department of Chemistry Towards the Synthesis of Isocoronene Iain William Currie This thesis is presented for the Degree of Doctor of Philosophy of Curtin University April 2018 Declaration To the best of my knowledge and belief this thesis contains no material previously published by any other person except where due acknowledgement has been made. This thesis contains no material which has been accepted for the award of any other degree or diploma in any other university. Signature: Date: i Abstract The concept of aromaticity and its implications are fundamentally important to a wide range of applied sciences involving organic molecules. Aromaticity arises from the delocalisation of electrons through a cyclic conjugated system known as a conjugated circuit. Monocyclic aromatic compounds possess a single conjugated circuit while polycyclic aromatic hydrocarbons (PAHs) may have numerous potential conjugated circuits. The aromaticity of PAHs is complicated by the presence of multiple conjugated circuits which may have varying contribution to the overall properties depending on several factors such as geometry and topology. Isocoronene 105 is one example of a PAH classified as a non-benzenoid corannulene. Isocoronene is unique among corannulenes since the conjugated circuits are restricted to the peripheral and central rings only. Isocoronene has been used as a model compound for computational studies into aromaticity and may provide the first example of a superaromatic molecule. The synthesis of novel aromatic structures such as isocoronene is essential in providing unambiguous empirical data which can be used to verify and develop computational methods. In addition, the development of new synthetic methodologies towards PAHs is important in the field of organic electronics. -

Decomposition of Ruthenium Olefin Metathesis Catalyst
catalysts Review Decomposition of Ruthenium OlefinOlefin Metathesis CatalystMetathesis Catalyst Magdalena Jawiczuk 1,, Anna Anna Marczyk Marczyk 1,21,2 andand Bartosz Bartosz Trzaskowski Trzaskowski 1,* 1,* 1 1 CentreCentre of of New New Technologies, Technologies, University University of of Warsaw, Warsaw, Banacha Banacha 2c, 2c, 02-097 02-097 Warsaw, Warsaw, Poland; [email protected]@cent.uw.edu.pl (M.J.); (M.J.); [email protected] [email protected] (A.M.) (A.M.) 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland 2 Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland * Correspondence: [email protected] * Correspondence: [email protected] Received: 28 28 June 2020; Accepted: 02 2 AugustAugust 2020;2020; Published:Published: 5date August 2020 Abstract: RutheniumRuthenium olefin olefin metathesis metathesis catalysts catalysts are are one one of of the most commonly used class of catalysts. There There are are multiple multiple reviews reviews on on their their us useses in in various branches of chemistry and other sciences but a detailed review of their decomposition is missing, despite a large number of recent and important advances advances in in this this field. field. In In particular, particular, in in the the last last five five years years several several new new mechanism mechanism of decomposition,of decomposition, both both olefin-driven olefin-driven as well as well as induc as induceded by external by external agents, agents, have have been been suggested suggested and usedand usedto explain to explain differences differences in the decomposition in the decomposition rates and rates the metathesis and the metathesis activities activitiesof both standard, of both N-heterocyclicstandard, N-heterocyclic carbene-based carbene-based systems and systems the recently and the developed recently developed cyclic alkyl cyclic amino alkyl carbene- amino containingcarbene-containing complexes.