World Journal of Pharmaceutical Research Rafik Et Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neurontin (Gabapentin)
Texas Prior Authorization Program Clinical Criteria Drug/Drug Class Gabapentin Clinical Criteria Information Included in this Document Neurontin (gabapentin) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. Gralise (gabapentin Extended Release) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. March 29, 2019 Copyright © 2019 Health Information Designs, LLC 1 Horizant -

Emerging Drug List PARECOXIB SODIUM
Emerging Drug List PARECOXIB SODIUM NO. 10 MAY 2001 Generic (Trade Name): Parecoxib sodium Manufacturer: Pharmacia & Upjohn Inc. Indication: For peri-operative pain relief Current Regulatory Parecoxib is currently under review at Health Canada and the Food and Drug Status: Administration in the U.S. They are expecting approval in the fourth quarter of 2001. It is not marketed in any country at this time Description: Parecoxib is the first parenteral cyclooxygenase-2 (COX-2) selective inhibitor to be developed. It is a water-soluble prodrug that is rapidly hydrolyzed to valdecoxib (the active COX-2 inhibitor). Valdecoxib's affinity for COX-2 versus COX-1 is 90 times greater than celecoxib and 34,000 times greater than ketorolac. Once injected, peak concentrations of valdecoxib are attained in 10 to 20 minutes. Valdecoxib has a half-life of eight to 10 hours. Current Treatment: Currently, the only other nonsteroidal anti-inflammatory agent that is available as an injection is ketorolac. Ketorolac is used in some centres for peri-operative pain management, however opioids are the main class of agents used for this indication. Ketorolac has been associated with a relatively high incidence of gastrointestinal (GI) side effects, including severe cases of hemorrhage. Cost: There is no information available on the cost of parecoxib. Evidence: Parecoxib has been compared to ketorolac and morphine in clinical trials. Parecoxib at a dose of 20 and 40 mg/day IV, were compared to ketorolac, 30 mg/day IV and morphine, 4 mg/day or placebo in 202 women undergoing hysterectomy. Both doses of parecoxib were comparable to ketorolac for relieving postoperative pain. -

Big Pain Assays Aren't a Big Pain with the Raptor Biphenyl LC Column
Featured Application: 231 Pain Management and Drugs of Abuse Compounds in under 10 Minutes by LC-MS/MS Big Pain Assays Aren’t a Big Pain with the Raptor Biphenyl LC Column • 231 compounds, 40+ isobars, 10 drug classes, 22 ESI- compounds in 10 minutes with 1 column. • A Raptor SPP LC column with time-tested Restek Biphenyl selectivity is the most versatile, multiclass-capable LC column available. • Achieve excellent separation of critical isobars with no tailing peaks. • Run fast and reliable high-throughput LC-MS/MS analyses with increased sensitivity using simple mobile phases. The use of pain management drugs is steadily increasing. As a result, hospital and reference labs are seeing an increase in patient samples that must be screened for a wide variety of pain management drugs to prevent drug abuse and to ensure patient safety and adherence to their medication regimen. Thera- peutic drug monitoring can be challenging due to the low cutoff levels, potential matrix interferences, and isobaric drug compounds. To address these chal- lenges, many drug testing facilities are turning to liquid chromatography coupled with mass spectrometry (LC-MS/MS) for its increased speed, sensitivity, and specificity. As shown in the analysis below, Restek’s Raptor Biphenyl column is ideal for developing successful LC-MS/MS pain medication screening methodologies. With its exceptionally high retention and unique selectivity, 231 multiclass drug compounds and metabolites—including over 40 isobars—can be analyzed in just 10 minutes. In addition, separate panels have been optimized on the Raptor Biphenyl column specifically for opioids, antianxiety drugs, barbiturates, NSAIDs and analgesics, antidepressants, antiepileptics, antipsychotics, hallucinogens, and stimulants for use during confirmation and quantitative analyses. -

Efficacy and Safety of Postoperative Intravenous Parecoxib Sodium
Open Access Protocol BMJ Open: first published as 10.1136/bmjopen-2016-011732 on 8 September 2016. Downloaded from Efficacy and safety of Postoperative Intravenous Parecoxib sodium Followed by ORal CElecoxib (PIPFORCE) post- total knee arthroplasty in patients with osteoarthritis: a study protocol for a multicentre, double-blind, parallel- group trial Qianyu Zhuang,1 Yanyan Bian,1 Wei Wang,1 Jingmei Jiang,2 Bin Feng,1 Tiezheng Sun,3 Jianhao Lin,3 Miaofeng Zhang,4 Shigui Yan,4 Bin Shen,5 Fuxing Pei,5 Xisheng Weng1 To cite: Zhuang Q, Bian Y, ABSTRACT Strengths and limitations of this study Wang W, et al. Efficacy and Introduction: Total knee arthroplasty (TKA) has been safety of Postoperative regarded as a most painful orthopaedic surgery. ▪ Intravenous Parecoxib This is the first study to investigate the efficacy Although many surgeons sequentially use parecoxib sodium Followed by ORal and safety of the sequential analgesia regimen of CElecoxib (PIPFORCE) post- and celecoxib as a routine strategy for postoperative intravenous parecoxib followed by oral celecoxib total knee arthroplasty in pain control after TKA, high quality evidence is still after total knee arthroplasty surgery. patients with osteoarthritis: a lacking to prove the effect of this sequential regimen, ▪ This study will explore the benefits of prolonged study protocol for a especially at the medium-term follow-up. The purpose sequential treatment of parecoxib and celecoxib multicentre, double-blind, of this study, therefore, is to evaluate efficacy and in medium-term function recovery. parallel-group trial. BMJ safety of postoperative intravenous parecoxib sodium ▪ The results will promote the non-steroidal Open 2016;6:e011732. -

Chapter 25 Mechanisms of Action of Antiepileptic Drugs
Chapter 25 Mechanisms of action of antiepileptic drugs GRAEME J. SILLS Department of Molecular and Clinical Pharmacology, University of Liverpool _________________________________________________________________________ Introduction The serendipitous discovery of the anticonvulsant properties of phenobarbital in 1912 marked the foundation of the modern pharmacotherapy of epilepsy. The subsequent 70 years saw the introduction of phenytoin, ethosuximide, carbamazepine, sodium valproate and a range of benzodiazepines. Collectively, these compounds have come to be regarded as the ‘established’ antiepileptic drugs (AEDs). A concerted period of development of drugs for epilepsy throughout the 1980s and 1990s has resulted (to date) in 16 new agents being licensed as add-on treatment for difficult-to-control adult and/or paediatric epilepsy, with some becoming available as monotherapy for newly diagnosed patients. Together, these have become known as the ‘modern’ AEDs. Throughout this period of unprecedented drug development, there have also been considerable advances in our understanding of how antiepileptic agents exert their effects at the cellular level. AEDs are neither preventive nor curative and are employed solely as a means of controlling symptoms (i.e. suppression of seizures). Recurrent seizure activity is the manifestation of an intermittent and excessive hyperexcitability of the nervous system and, while the pharmacological minutiae of currently marketed AEDs remain to be completely unravelled, these agents essentially redress the balance between neuronal excitation and inhibition. Three major classes of mechanism are recognised: modulation of voltage-gated ion channels; enhancement of gamma-aminobutyric acid (GABA)-mediated inhibitory neurotransmission; and attenuation of glutamate-mediated excitatory neurotransmission. The principal pharmacological targets of currently available AEDs are highlighted in Table 1 and discussed further below. -
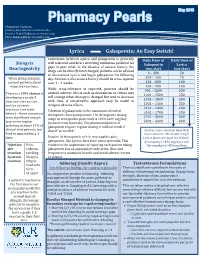
Lyrica Gabapentin: an Easy Switch!
Pharmacist Contacts: [email protected]; [email protected]; [email protected] Lyrica Gabapentin: An Easy Switch! Conversion between Lyrica and gabapentin is generally Daily Dose of Daily Dose of Shingrix well tolerated and direct switching minimizes potential for Gabapentin Lyrica Reactogenicity gaps in pain relief. In the absence of seizure history, the (mg/day) (mg/day) drugs can be directly interchanged; patients can be advised 0 – 300 50 to discontinue Lyrica and begin gabapentin the following When giving Shingrix, day. Patients with a seizure history should be cross-tapered 301 – 450 75 counsel patients about over 1 – 4 weeks. 451 – 600 100 expected reactions. 601 – 900 150 While cross-tolerance is expected, patients should be 901 – 1200 200 advised adverse effects such as drowsiness or edema may There is a 10% chance of 1201 – 1500 250 still emerge when therapy is changed but tend to decrease developing a grade 3 1501 – 1800 300 injection site reaction with time. A conservative approach may be useful to 1801 – 2100 350 and/or systemic mitigate adverse effects. reactions (see table 2101 – 2400 400 Titration of gabapentin to the maximum tolerated 2401 – 2700 450 below) – these symptoms therapeutic dose is important. The therapeutic dosing 2701 – 3000 500 were significant enough range in neuropathic pain trials is 1800-3600 mg/day to prevent regular (normal renal function). The pharmacokinetics of 3001 – 3600 600 activities in about 17% of gabapentin require regular dosing, it will not work if clinical trial patients, but dosed “as needed.” Studies show minimal benefit & tend to pass within 2-3 more adverse effects when high days. -

Comparing Adverse Effects of Analgesic Strategies For
Comparing Adverse Effects daily and weekly time during which higher than that of the only ATC opi- their pain interfered with their mood oids group and 17 times higher than of Analgesic Strategies for or activities, and they indicated the that of the only PRN opioids group. Chronic Cancer Pain amount of relief they received from Source: J Pain Symptom Manage. 2007;33(1):67–77. their pain medicine in the previ- Nausea, vomiting, drowsiness, lack ous week. They also completed the of energy, urinary retention—some- Karnofsky Performance Status (KPS), Clopidogrel: Best Results times the adverse effects of the anal- which measures patients’ ability to per- Pre- or Post-PCI? gesic medication are enough to derail form activities of daily living and their pain management for patients with need for caregiver assistance. When is the best time to give the anti- cancer. It would help to understand There were no significant differ- platelet clopidogrel to patients sched- the relationships between the type of ences between any of the four groups uled for diagnostic coronary angiogra- analgesic prescription and the preva- in terms of pain intensity or amount of phy—before ad hoc coronary stenting lence and severity of adverse effects. time spent in pain. Total pain interfer- or immediately after? Researchers from But not much information is available, ence scores, however, were significantly University of Debrecen, Debrecen, say researchers from the University higher in the ATC plus PRN opioids Hungary and Medical University of of California, San Francisco; the Uni- group than in the no opioids group. Vienna and Wilhelminenhospital, both versity of Nebraska, Omaha; and the The ATC plus PRN opioids group in Vienna, Austria say starting treat- University of Texas Southwestern Med- also had significantly lower functional ment before percutaneous coronary ical Center, Dallas. -

Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review
Current Pain and Headache Reports (2019) 23: 37 https://doi.org/10.1007/s11916-019-0774-0 OTHER PAIN (A KAYE AND N VADIVELU, SECTION EDITORS) Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review Omar Viswanath1,2,3 & Ivan Urits4 & Mark R. Jones4 & Jacqueline M. Peck5 & Justin Kochanski6 & Morgan Hasegawa6 & Best Anyama7 & Alan D. Kaye7 Published online: 1 May 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose of Review Neuropathic pain is often debilitating, severely limiting the daily lives of patients who are affected. Typically, neuropathic pain is difficult to manage and, as a result, leads to progression into a chronic condition that is, in many instances, refractory to medical management. Recent Findings Gabapentinoids, belonging to the calcium channel blocking class of drugs, have shown good efficacy in the management of chronic pain and are thus commonly utilized as first-line therapy. Various sodium channel blocking drugs, belonging to the categories of anticonvulsants and local anesthetics, have demonstrated varying degrees of efficacy in the in the treatment of neurogenic pain. Summary Though there is limited medical literature as to efficacy of any one drug, individualized multimodal therapy can provide significant analgesia to patients with chronic neuropathic pain. Keywords Neuropathic pain . Chronic pain . Ion Channel blockers . Anticonvulsants . Membrane stabilizers Introduction Neuropathic pain, which is a result of nervous system injury or lives of patients who are affected. Frequently, it is difficult to dysfunction, is often debilitating, severely limiting the daily manage and as a result leads to the progression of a chronic condition that is, in many instances, refractory to medical This article is part of the Topical Collection on Other Pain management. -

Insomnia Disorder a VA Clinician’S Guide to Managing Insomnia Disorder (2019) Contents Insomnia Disorder
Insomnia Disorder A VA Clinician’s Guide to Managing Insomnia Disorder (2019) Contents Insomnia Disorder .................................................................................................... 3 Risks in elderly patients and patients with dementia .................................15 Background ................................................................................................................ 3 Provider perceptions vs reality ............................................................................16 Figure 1. Stepped Care for Management of Insomnia Disorder .............. 3 Figure 5. Weighing the potential risks versus benefits of Table 1. Brief summary of the ISI ......................................................................... 4 medication use .........................................................................................................16 Figure 2. Acute Insomnia to Insomnia Disorder ............................................ 5 Doxepin ........................................................................................................................17 Clinical Pearl ............................................................................................................... 5 Figure 6. Doxepin Use .............................................................................................18 Figure 3. Common causes of sleep disturbance ........................................... 6 Ramelteon ...................................................................................................................18 -

Safety of Etoricoxib in Patients with Reactions to Nsaids
ORIGINAL ARTICLE Safety of Etoricoxib in Patients With Reactions to NSAIDs O Quercia, F Emiliani, FG Foschi, GF Stefanini Allergology High Specialty Unit, General Medicine, Faenza Hospital, AUSL Ravenna, Italy ■ Abstract Background: Adverse reactions to nonsteroidal anti-infl ammatory drugs (NSAIDs) are a frequently reported problem due to the fact that these molecules are often used for control of pain and infl ammation. Although the use of selective inhibitors of cyclooxygenase (COX) 2 helps to prevent some of these adverse reactions, they can have cardiac side effects when taken for prolonged periods. Here we report the safety and tolerability of etoricoxib, a selective COX-2 inhibitor with fewer cardiovascular effects, in patients with adverse reactions to NSAIDs. Patients and methods: We performed placebo-controlled oral challenge with etoricoxib in 65 patients with previous adverse reactions to NSAIDs: 13 to salicylates, 18 to arylpropionic acids, 10 to arylacetic acid, 12 to oxicam and derivates, 8 to pyrazolones, and 4 to acetaminophen (paracetamol). The reported symptoms were urticaria or angioedema in 69%, rhinitis in 3%, and 1 case of anaphylactic shock (1.5%). The challenge was done using the placebo on the fi rst day, half dosage of etoricoxib (45 mg) on the second day, and the therapeutic dose of 90 mg on the third day. The challenge was done in the outpatient department of the hospital and the subjects were monitored for a further 4 to 6 hours after challenge. Results: Oral challenge with etoricoxib was well tolerated in 97% of the patients. Only 2 systemic reactions were reported during the challenge test. -

1 Gabapentinoids in Total Joint Arthroplasty: the Clinical Practice
1 Gabapentinoids in Total Joint Arthroplasty: The Clinical Practice Guidelines of the American Association of Hip and Knee Surgeons, American Society of Regional Anesthesia and Pain Medicine, American Academy of Orthopaedic Surgeons, Hip Society, and Knee Society Charles P. Hannon MD1, Yale A. Fillingham MD2, James A. Browne MD3, Emil H Schemitsch MD FRCS(C)4, AAHKS Anesthesia & Analgesia Clinical Practice Guideline Workgroup5, Asokumar Buvanendran MD6, William G. Hamilton MD7*, Craig J. Della Valle MD1* 1Department of Orthopaedic Surgery, Rush University Medical Center, Chicago, IL, USA 2Department of Orthopaedic Surgery, Dartmouth-Hitchcock Medical Center, Lebanon, NH, USA 3Department of Orthopaedic Surgery, University of Virginia, Charlottesville, VA, USA 4Department of Surgery, University of Western Ontario, London, Ontario, Canada 5Workgroup Comprised of the following individuals: Justin T. Deen MD (Department of Orthopaedics and Rehabilitation, University of Florida College of Medicine, Gainesville, FL, USA), Greg A. Erens MD (Department of Orthopaedic Surgery, Emory University, Atlanta, GA, USA), Jess H. Lonner MD (Rothman Institute at Thomas Jefferson University, Philadelphia, PA, USA), Aidin E. Pour MD (Department of orthopaedic surgery, University of Michigan, Ann Arbor, MI, USA), Robert S. Sterling MD (Department of Orthopedic Surgery, Johns Hopkins University School of Medicine, Baltimore, MD, USA) 6Department of Anesthesiology, Rush University Medical Center, Chicago, IL, USA 7Anderson Orthopedic Research Institute, Alexandria, VA, USA *Denotes co-senior authors 2 Introduction The American Association of Hip and Knee Surgeons (AAHKS), The American Academy of Orthopaedic Surgeons (AAOS), The Hip Society, The Knee Society and The American Society of Regional Anesthesia and Pain Medicine (ASRA) have worked together to develop evidence-based guidelines on the use of gabapentinoids in primary total joint arthroplasty (TJA). -

Use of Parecoxib by Continuous Subcutaneous Infusion for Cancer Pain in a Hospice Population
BMJ Support Palliat Care: first published as 10.1136/bmjspcare-2017-001348 on 1 September 2017. Downloaded from Short report Use of parecoxib by continuous subcutaneous infusion for cancer pain in a hospice population Peter Armstrong,1 Pauline Wilkinson,2 Noleen K McCorry3 1Pharmacy Department, Belfast ABSTRACT adverse effects.2 Parecoxib, a prodrug Health and Social Care Trust, Objectives To characterise the use of the of valdecoxib, is an injectable selective Belfast, UK 2Consultant in Palliative parenteral non-steroidal anti-inflammatory COX-2 inhibitor marketed in the UK as Medicine, Marie Curie Hospice, drug parecoxib when given by continuous Dynastat Injection by Pfizer Limited. It Belfast, UK subcutaneous infusion (CSCI) in a hospice is licensed for the short-term treatment 3 Centre of Excellence for Public population. Clinical experience suggests of postoperative pain in adults by the Health (NI), Queen’s University 3 Belfast, Belfast, UK parecoxib CSCI may be of benefit in this intramuscular or intravenous routes. It population, but empirical evidence in relation to has been used extensively by continuous Correspondence to its safety and efficacy is lacking. subcutaneous infusion (CSCI) in pallia- Mr. Peter Armstrong; Methods Retrospective chart review of patients peter. armstrong@ belfasttrust. tive care in Northern Ireland; however, hscni. net with a cancer diagnosis receiving parecoxib CSCI evidence for its safety and efficacy by this from 2008 to 2013 at the Marie Curie Hospice, route is lacking. Received 3 March 2017 Belfast. Data were collected on treatment Revised 9 August 2017 Accepted 16 August 2017 regime, tolerability and, in patients receiving AIM OF THE STUDY at least 7 days treatment, baseline opioid dose This study aims to: and changes in pain scores or opioid rescue 1.