Computational Identification of Regulatory Features Affecting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
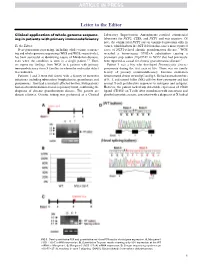
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

The Use of Phosphoproteomic Data to Identify Altered Kinases and Signaling Pathways in Cancer
The use of phosphoproteomic data to identify altered kinases and signaling pathways in cancer By Sara Renee Savage Thesis Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Biomedical Informatics August 10, 2018 Nashville, Tennessee Approved: Bing Zhang, Ph.D. Carlos Lopez, Ph.D. Qi Liu, Ph.D. ACKNOWLEDGEMENTS The work presented in this thesis would not have been possible without the funding provided by the NLM training grant (T15-LM007450) and the support of the Biomedical Informatics department at Vanderbilt. I am particularly indebted to Rischelle Jenkins, who helped me solve all administrative issues. Furthermore, this work is the result of a collaboration between all members of the Zhang lab and the larger CPTAC consortium. I would like to thank the other CPTAC centers for processing the data, and Chen Huang and Suhas Vasaikar in the Zhang lab for analyzing the colon cancer copy number and proteomic data, respectively. All members of the Zhang lab have been extremely helpful in answering any questions I had and offering suggestions on my work. Finally, I would like to acknowledge my mentor, Bing Zhang. I am extremely grateful for his guidance and for giving me the opportunity to work on these projects. ii TABLE OF CONTENTS Page ACKNOWLEDGEMENTS ................................................................................................ ii LIST OF TABLES............................................................................................................