2Nd Postdoc Methods Symposium Program
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2010-2011 Annual Report
Annual Report 2010-2011 Mastery of disease through discovery | www.wehi.edu.au Contents 1 About the institute 3 Director’s and Chairman’s report 5 Discovery 8 Cancer and Haematology 10 Stem Cells and Cancer 12 Molecular Genetics of Cancer 14 Chemical Biology 16 Molecular Medicine 18 Structural Biology 20 Bioinformatics 22 Infection and Immunity 24 Immunology The Walter and Eliza Hall Institute 26 Autoimmunity and Transplantation of Medical Research 28 Cell Signalling and Cell Death 1G Royal Parade 30 Inflammation Parkville Victoria 3052 Australia Telephone: (+61 3) 9345 2555 32 Molecular Immunology Facsimile: (+61 3) 9347 0852 34 Publications WEHI Biotechnology Centre 36 Awards 4 Research Avenue 37 Translation La Trobe R&D Park Bundoora Victoria 3086 Australia Translating our research 38 Telephone: (+61 3) 9345 2200 40 Developing our research Facsimile: (+61 3) 9345 2211 42 Patents www.wehi.edu.au www.facebook.com/WEHIresearch 43 Education www.twitter.com/WEHI_research 46 2010-11 graduates ABN 12 004 251 423 47 Seminars Acknowledgements 48 Institute awards Produced by the institute’s Community Relations department 49 Engagement Managing editor: Penny Fannin Editor: Liz Williams 51 Strategic partners Writers: Liz Williams, Vanessa Solomon and Julie Tester 52 Scientific and medical community Design and production: Simon Taplin Photography: Czesia Markiewicz and Cameron Wells 54 Public engagement 57 Engagement with schools Cover image 58 Donor and bequestor engagement Art in Science finalist 2010 Vessel webs 59 Sustainability Dr Leigh Coultas, Cancer and Haematology division 60 The Board This image shows the delicate intricacy in the developing eye of a transient population of web-like blood vessels. -

Survey of Commercial Outcomes from Public Research (Scopr) 2019 Report
techtransfer.org.au SURVEY OF COMMERCIAL OUTCOMES FROM PUBLIC RESEARCH (SCOPR) 2019 REPORT Survey and report delivered by FOREWORD There is an ever-present imperative to capture the commercial value of our research endeavour for our future wellbeing. To do so strategically, decision makers from laboratory, institutional and government levels need insights into how the research sector is currently engaging with industry to transfer knowledge and innovation, and thereby deliver benefits to our society from the fruits of our research. For many years in Australia there has been a focus on improving innovation metrics, thus I am delighted to acknowledge the initiative of gemaker and Knowledge Commercialisation Australasia (KCA) in producing the inaugural Survey of Commercial Outcomes from Public Research (SCOPR). The SCOPR takes its lead from the National Survey of Research Commercialisation (NSRC) produced since 2000 by the Department of Industry, Science, Energy and Resources. To avoid duplication, the Department has decided to cease the NSRC and will work with KCA to share knowledge, and access data collected by SCOPR. As we face the COVID-19 pandemic, effective knowledge transfer is more important than ever, so I hope that this report will spur our research institutions to even greater achievements. Realising effective knowledge transfer will depend on having skilled commercialisation professionals who can help researchers turn great ideas into beneficial products and services. I applaud KCA’s support for technology transfer professionals -

The Bio21 Institute's 2020 Annual Report Is Available to Download
Annual Report 2020 Image of the coronavirus SARS-CoV-2 taken by Andrew Leis and Jason Roberts. Courtesy of the Doherty Institute. The Bio21 Molecular Science and Director Associate Director – Platform Biotechnology Institute Professor Michael W. Parker Infrastructure University of Melbourne DPhil (Oxon) FAA FAHMS Professor Malcolm McConville PhD 30 Flemington Road Deputy Director Associate Director – Commercialisation Parkville Victoria 3010 Professor Frances Separovic AO Professor Spencer Williams PhD Telephone: (03) 8344 2220 PhD FAA www.bio21.unimelb.edu.au Associate Director – Engagement @Bio21Institute Professor Sally Gras PhD @Bio21Institute Scientific Research Manager Dr David Keizer, PhD Produced by the Bio21 Molecular Science and Biotechnology External Relations Advisor,b Bio21 Florienne Institute Loder Annual Report 2020 Contents Our Mission 2 Our Vision 2 About the Institute 3 Director’s Message 4 Bio21 Leadership 8 Deputy Director, Professor Emeritus Frances Separovic AO 8 Associate Director Engagement – Professor Sally Gras 10 Associate Director Commercialisation – Professor Spencer Williams 13 Associate Director Platform Infrastructure – Professor Malcolm McConville 14 ACRF Facility for Innovative Cancer Drug Discovery 16 Impacts of Research 19 OHS Report 31 Equity Diversity and Inclusion 32 Industry Engagement and Commercialisation 35 Announcing the Ruth Bishop Building and Ian Holmes Imaging Centre 36 Events and Conferences 39 Graduate Research Students and Early Career Researchers 41 Institute Members Honoured 42 Grant Successes 43 Governance 46 Bio21 Scientific Research Team 47 Bio21 Research Groups 48 Bio21 People 50 Institute in Numbers 56 Bio21 Institute Theses submitted in 2020 57 Bio21 Steering Committee 58 Industry partners 63 Produced by the Bio21 Molecular Science aFontn Front cover image: Bio21 precinct aerial photograph, courtesy of Kane Jarrod Photography. -
2Nd Postdoc Methods Symposium Program
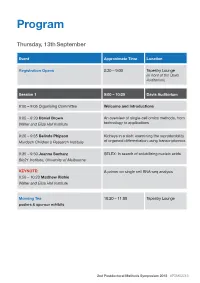
Program Thursday, 13th September Event Approximate Time Location Registration Opens 8:30 – 9:00 Tapestry Lounge (in front of the Davis Auditorium) Session 1 9:00 – 10:20 Davis Auditorium 9:00 – 9:05 Organising Committee Welcome and Introductions 9:05 – 9:20 Daniel Brown An overview of single-cell omics methods, from Walter and Eliza Hall Institute technology to applications 9:20 – 9:35 Belinda Phipson Kidneys in a dish: examining the reproducibility Murdoch Children’s Research Institute of organoid differentiation using transcriptomics 9:35 – 9:50 Joanna Sacharz SELEX: In search of solubilizing nucleic acids Bio21 Institute, University of Melbourne KEYNOTE: A primer on single cell RNA-seq analysis 9:50 – 10:20 Matthew Richie Walter and Eliza Hall Institute Morning Tea 10:20 – 11:00 Tapestry Lounge posters & sponsor exhibits 2nd Postdoctoral Methods Symposium 2018 #PDMS2018 Event Approximate Time Location Session 2 11:00 – 12:15 Davis Auditorium MCFP sponsored talk Biological Imaging via Helium ion Microscopy 11:00 – 11:15 Babak Nasr University of Melbourne 11:15 – 11:30 Mohamed Fareh Single-molecule fluorescence reveals the Peter MacCallum Cancer Centre dynamics of microRNA recognition by Dicer- TRBP complex 11:30 – 11:45 Charis Teh Capturing the cellular gymnastics of survival Walter and Eliza Hall institute and killer proteins by mass cytometry (CyTOF) 11:45 – 12:00 Jieqiong Lou Phasor analysis and image correlation Bio21 Institute, University of Melbourne spectroscopy of histone FLIM/FRET reveals spatiotemporal regulation of chromatin organization by the DNA damage response. Session 2 Flash talks 12:00 – 12:15 Davis Auditorium Rachel Lundie Flow FISH as a method to elucidate the Walter and Eliza Hall Institute transcriptional effects of M. -

Drug Discovery and Technology Capabilities at WEHI Melbourne, Australia History of WEHI
Drug Discovery and Technology Capabilities at WEHI Melbourne, Australia History of WEHI • Established 1915 • Focused on the fundamental principles of medical biology to mitigate disease • Independent Research Institute affiliated with University of Melbourne, Royal Melbourne Hospital and the Victorian Comprehensive Cancer Center WEHI Overview 90 laboratories working on 120 Clinical trials 50+ diseases currently underway > +30 million patients world wide have 1200+ researchers benefited from our research and staff World-Class Research Across a Broad Range of Therapeutic Areas • WEHI performs influential basic and translational research focused on four key therapeutic areas: – Cancer – Immunology & Inflammation – Infectious diseases – Ageing and development Font size proportional to research effort per topic Fulfilling Our Mission • WEHI was ranked 19th in the 2019 Nature Index for global not-for-profit/non-governmental organizations. • Translating discoveries: Venetoclax = New targeted treatment for Chronic Lymphocytic Leukaemia approved by FDA in 2016. • Gender Equity in Research: WEHI is recipient of SAGE Athena SWAN Award + membership of Male Champions of Change. • Increased funding partnership with State government ➣ State government invests $18M in WEHI National Drug Discovery Centre (in addition to contributions announced by Federal government). History of our Drug Discovery Centre • First academic drug discovery infrastructure at an Australian research institute – High-throughput screening facility – Medicinal chemistry laboratories -

2020 Annual Report
2020 Annual Report Make this cover come alive with augmented reality. Details on inside back cover. Contents The Walter and Eliza Hall Institute About WEHI 1 of Medical Research President’s report 2 Parkville campus 1G Royal Parade Director’s report 3 Parkville Victoria 3052 Australia Telephone: +61 3 9345 2555 WEHI’s new brand launched 4 Bundoora campus 4 Research Avenue Our supporters 10 La Trobe R&D Park Bundoora Victoria 3086 Australia Exceptional science and people 13 Telephone: +61 3 9345 2200 www.wehi.edu.au 2020 graduates 38 WEHIresearch Patents granted in 2020 40 WEHI_research WEHI_research WEHImovies A remarkable place 41 Walter and Eliza Hall Institute Operational overview 42 ABN 12 004 251 423 © The Walter and Eliza Hall Institute Expanding connections with our alumni 45 of Medical Research 2021 Diversity and inclusion 46 Produced by the WEHI’s Communications and Marketing department Working towards reconciliation 48 Director Organisation and governance 49 Douglas J Hilton AO BSc Mon BSc(Hons) PhD Melb FAA FTSE FAHMS WEHI Board 50 Deputy Director, Scientific Strategy WEHI organisation 52 Alan Cowman AC BSc(Hons) Griffith PhD Melb FAA FRS FASM FASP Members of WEHI 54 Chief Operating Officer WEHI supporters 56 Carolyn MacDonald BArts (Journalism) RMIT 2020 Board Subcommittees 58 Chief Financial Officer 2020 Financial Statements 59 Joel Chibert BCom Melb GradDipCA FAICD Financial statements contents 60 Company Secretary Mark Licciardo Statistical summary 94 BBus(Acc) GradDip CSP FGIA FCIS FAICD The year at a glance 98 Honorary -

Volunteer Blood Donor Registry Coordinator
Position description Volunteer Blood Donor Registry Coordinator Position title: Volunteer Blood Donor Registry Work location: RMH and WEHI, Parkville Coordinator Division/Department: Clinical Translation Centre Employment type: One year part-time (0.4FTE) with (RMH) and Clinical Translation (WEHI) potential for extension Position reference: WEHI/ANLL Further information: Lina Laskos [email protected] 03 9345 2304), Jo Casey [email protected] 03 9345 2480 Remuneration range: Closing date: Classification: HEW 6 – 7, dependent on experience Position reports to: Joint Heads, Clinical Translation (WEHI) and CTC Manager (RMH) Positions reporting to this one: Nil Position overview The role of the Volunteer Blood Donor Registry (VBDR) Coordinator is to manage and coordinate the activities of a well-established blood registry. The Volunteer Blood Donor Registry is a not-for-profit service established by the Walter and Eliza Hall Institute of Medical Research (WEHI) to support medical research in the Parkville precinct that requires the use of human blood. Registered healthy volunteers donate a small volume of blood for use in ethically approved research projects to facilitate clinical research. This position will coordinate the VBDR and support its transition from WEHI to Melbourne Health (MH), where it will operate under the auspices of the MH Clinical Trials Centre (CTC), as a joint initiative with WEHI. The VBDR Coordinator will organise current donors, consent and data collection in accordance with approved protocols, whilst maintaining confidentiality at all times. The VBDR Coordinator will regularly liaise with laboratory researchers about accessing the service, maintain the VBDR database and coordinate ethics, governance and reporting requirements. -

Position Description Template
Position description Senior Research Computing Engineer Position title: Senior Research Computing Scientist Classification: HEW 8 – HEW 9 Division/Department: Computational Biology Theme Work location: Parkville Position reference: WEHI/DASRCE250620 Employment type: 2 yr full time contract Remuneration range: Further information: Evan Thomas [email protected] 0423 000 246 Position reports to: Head of the Research Computing Closing date: Platform Positions reporting to this one: NA Position overview The Institute is committed to ongoing investment to provide researchers with access to state of the art computational and data capabilities, new technologies and expertise. Having implemented and continuing to grow the Institute’s private HPC cloud, new roles have been created to ensure that researchers can make the most of WEHI facilities and external research and cloud infrastructure. This includes roles to provide software infrastructure support services to enable high quality, computationally and data intensive research. This is a science enablement role the purpose of which is to develop solutions for computational science in complex enviroments spanning traditional on-premise facilities to cloud and complex hybrid execution models. The role requires development of productive relationships with researchers and IT staff to lead development in areas such as pipeline and pipeline tool development, data management and data movement tools, database development, machine learning pipelines, collaborative planning of hardware and software infrastructure, etc. In addition, the incumbent will work with IT and researchers to develop policy and guide regular upgrades to our compute infrastructure. The successful candidate will have will have an excellent understanding of computational research and the needs of researchers, excellent communication and interpersonal skills, and strong software engineering experience in an HPC environment. -

Position Description Template
Position description Research Tissue Coordinator Position title: Research Tissue Coordinator Classification: HEW 6 Division/Department: Gibbs Lab (Personalised Work location: Parkville campus + Western Health Oncology Division) (Sunshine/Footscray Campus) Position reference: WEHI/ANME Employment type: 1 year Full time or Part Time contract Remuneration range: Further information: Maria Edmonds - [email protected] Position reports to: Laboratory Manager Gibbs Closing date: 9 August 2019 Lab/Breast Cancer Laboratory and Melbourne Health Tissue Bank Manager Leanne Taylor Positions reporting to this one: Nil Position overview The Walter and Eliza Hall Institute of Medical Research (WEHI) has a number of divisions in Cancer Research (Blood Cells and blood cancer, Cancer biology and stem cells, and Personalised oncology). These positions will be supporting the Cancer Research projects which will be undertaken within this division at WEHI as well as other collaborative Victorian Comprehensive Cancer Centre (VCCC) studies. The newly created positions are a part of the Institutes’ strategic initiative to build relationships with Western Hospital and expand on the current level of service that Victorian Cancer Biobank (VCB) provides for the Western Hospital. The positions will also will work under the auspices of and towards the goals of the VCCC Understanding Response and Resistance to Targeted Therapies Program as outlined in the VCCC Strategic Research Plan (2017-2020). The Melbourne Health Tissue Bank operates in collaboration with The Victorian Cancer Biobank (VCB) as an open-access, not-for-profit tissue resource supported by The Cancer Council Victoria and the Victorian Government. The VCB provides cancer researchers with a diverse selection of high-quality bio-specimens and derivatives, comprehensively annotated with de-identified clinical outcome data. -

Submission to the Review of Independent Medical Research Institutes December 2014
Submission to the Review of Independent Medical Research Institutes December 2014 The Cooperative Research Centres Association represents all Australian Cooperative Research Centres (CRCs). In addition, the Association has universities, companies and research groups as Affiliate and Associate Members. Membership of the Association is optional for CRCs. The Association promotes best practice in research and translation; student supervision and contract management. For further inquiries contact: Dr. Tony Peacock Chief Executive Officer CRC Association 1/10 Bourke Street BARTON ACT 2600 02 6273 0624 www.crca.asn.au This work (Cooperative Research Centres Association 2014 - Submission to the Review of Independent Medical Research Institute A.J. Peacock) is free of known copyright restrictions. SUBMISSION TO THE REVIEW OF INDEPENDENT MEDICAL RESEARCH INSTITUTES Thank you for the opportunity to comment on the review. The Cooperative Research Centres (CRC) Association represents all of Australia’s Cooperative Research Centres as well as some Affiliate and Associate members. The CRC Programme is small in comparison to some other sources of medical research funding in Australia, but it is nevertheless an important source of funding. Currently there are eleven health and medical CRCs (Attachment A). The Government classifies the medical CRCs under “services” so it is not always clear what constitutes a medical CRC. By way of example, I have added the Capital Markets CRC as an 11th to the list. The Capital Markets CRC has worked extremely successfully on financial market integrity for almost two decades. Its recent renewal has seen it take a large focus on the $140 billion health market and is enjoy excellent support. -

Association of Australian Medical Research Institutes
SUBMISSION RESPONSE TO THE ISSUES PAPER SOUTH AUSTRALIAN PRODUCTIVITY COMMISSION INQUIRY INTO HEALTH AND MEDICAL RESEARCH IN SOUTH AUSTRALIA 18 May 2020 Contact: Professor Jonathan Carapetis AM President Association of Australian Medical Research Institutes PO Box 2097 Royal Melbourne Hospital VIC 3050 [email protected] www.aamri.org.au ABN 12 144 783 728 Contents About AAMRI .......................................................................................................................................... 3 Executive Summary ................................................................................................................................. 4 1 Medical research institutes: improving health outcomes through research ................................. 5 1.1 Australia’s MRIs: who they are and what they do .................................................................. 5 1.2 South Australia’s medical research institutes ......................................................................... 8 2 Considerations for assessing health and medical research performance and activity................... 9 2.1 Limitations of the proposed measures ................................................................................... 9 2.2 Expanding measures of health and medical research productivity and impact ................... 10 2.2.1 Measuring performance of health and medical research ............................................ 10 2.2.2 New framework for measuring impact of health and medical research ..................... -

Melbourne Knowledge Precincts
1 CITY/BIOMEDICAL PRECINCT > Aikenhead Centre for Medical Discovery MELBOURNE > Bio21 Institute > Bionics Institute > Centre for Eye Research Australia (CERA) KNOWLEDGE > Commonwealth Serum Laboratories (CSL) > CSIRO Materials Science and Engineering Parkville > IBM Research Collaboratory for Life Sciences PRECINCTS > Institute for a Broadband Enabled Society (IBES) > Ludwig Institute for Cancer Research > Melbourne Brain Centre > Mental Health Research Institute > National Ageing Research Institute (NARI) > National ICT Australia: Victorian Research Laboratory > O’Brien Institute of Microsurgery Melbourne is home to three knowledge > Orygen Youth Health Centre precincts bringing together academic > Peter Doherty Institute for Infection and Immunity institutions, research and medical > Peter MacCallum Cancer Centre > Royal Melbourne Institute of Technology (RMIT) centres and industry to foster world- > St Vincent’s Institute of Medical Research and leading innovation. St Vincent’s Health > Super Computer Initiative > The Royal Children’s Hospital > The Royal Melbourne Hospital > The Royal Women’s Hospital > University of Melbourne NORTHERN PRECINCT > Victorian Comprehensive Cancer Centre (VCCC) - Opens in 2015 > Victorian Life Sciences Computation Initiative (VLSCI) 3 > Walter and Eliza Hall Institute of Medical Research (WEHI) 2 SOUTH EAST MELBOURNE INNOVATION PRECINCT (SEMIP) CITY/BIOMEDICAL PRECINCT > Australian Synchrotron > Commonwealth Scientific and Industrial Research 1 Organisation (CSIRO) > Melbourne Centre for Nanofabrication