Guidelines on the Use of Therapeutic Apheresis in Clinical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recombinant Factors for Hemostasis
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Chemical & Biomolecular Engineering Theses, Chemical and Biomolecular Engineering, Dissertations, & Student Research Department of Summer 2010 Recombinant Factors for Hemostasis Jennifer Calcaterra University of Nebraska at Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/chemengtheses Part of the Biochemical and Biomolecular Engineering Commons Calcaterra, Jennifer, "Recombinant Factors for Hemostasis" (2010). Chemical & Biomolecular Engineering Theses, Dissertations, & Student Research. 5. https://digitalcommons.unl.edu/chemengtheses/5 This Article is brought to you for free and open access by the Chemical and Biomolecular Engineering, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Chemical & Biomolecular Engineering Theses, Dissertations, & Student Research by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Recombinant Factors for Hemostasis by Jennifer Calcaterra A DISSERTATION Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Doctor of Philosophy Major: Interdepartmental Area of Engineering (Chemical & Biomolecular Engineering) Under the Supervision of Professor William H. Velander Lincoln, Nebraska August, 2010 Recombinant Factors for Hemostasis Jennifer Calcaterra, Ph.D. University of Nebraska, 2010 Adviser: William H. Velander Trauma deaths are a result of hemorrhage in 37% of civilians and 47% military personnel and are the primary cause of death for individuals under 44 years of age. Current techniques used to treat hemorrhage are inadequate for severe bleeding. Preliminary research indicates that fibrin sealants (FS) alone or in combination with a dressing may be more effective; however, it has not been economically feasible for widespread use because of prohibitive costs related to procuring the proteins. -

Factors Affecting Mobilization of Peripheral Blood Progenitor Cells in Patients with Lymphoma’
Vol. 4, 311-316, February 1998 Clinical Cancer Research 311 Factors Affecting Mobilization of Peripheral Blood Progenitor Cells in Patients with Lymphoma’ Craig H. Moskowitz,2 Jill R. Glassman, (median, 13 versus 22 days; P 0.06). Patients who received 1l cycles of chemotherapy prior to PBPC mobilization David Wuest, Peter Maslak, Lilian Reich, tended to have delayed platelet recovery to >20,090/&l and Anthony Gucciardo, Nancy Coady-Lyons, to require more platelet transfusions than less extensively Andrew D. Zelenetz, and Stephen D. Nimer pretreated patients (median, 13.5 versus 23.5 days; P 0.15; Division of Hematologic Oncology, Department of Medicine median number of platelet transfusion episodes, 13 versus 9; [C. H. M., D. W., P. M., L. R., A. G., N. C-L., A. D. Z., S. D. N.] and P = 0.17). Department of Biostatistics [J. R. G.], Memorial Sloan-Kettering Cancer Center, New York, New York 10021 These data suggest that current strategies to mobilize PBPCs may be suboptimal in patients who have received either stem cell-toxic chemotherapy or 11 cycles of chem- ABSTRACT otherapy prior to PBPC mobilization. Alternative ap- The objective of this study was to identify factors asso- proaches, such as ex vivo expansion or the use of other ciated with poor mobilization of peripheral blood progenitor growth factors in addition to G-CSE, may improve mobili- cells (PBPCs) or delayed platelet engraftment after high- zation of progenitor cells for PBPC transplantation. dose therapy and autologous stem cell transplantation in patients with lymphoma. INTRODUCTION Fifty-eight patients with Hodgkin’s disease or non- The use of high-dose chemoradiotherapy supported by Hodgkin’s lymphoma underwent PBPC transplantation as cryopreserved autologous hematopoietic progenitor cells is ef- the “best available therapy” at Memorial Sloan-Kettering fective in treating relapsed HD3 and NHL; a high complete Cancer Center (New York, NY) between 1993 and 1995. -

Hemolytic Disease of the Newborn
Intensive Care Nursery House Staff Manual Hemolytic Disease of the Newborn INTRODUCTION and DEFINITION: Hemolytic Disease of the Newborn (HDN), also known as erythroblastosis fetalis, isoimmunization, or blood group incompatibility, occurs when fetal red blood cells (RBCs), which possess an antigen that the mother lacks, cross the placenta into the maternal circulation, where they stimulate antibody production. The antibodies return to the fetal circulation and result in RBC destruction. DIFFERENTIAL DIAGNOSIS of hemolytic anemia in a newborn infant: -Isoimmunization -RBC enzyme disorders (e.g., G6PD, pyruvate kinase deficiency) -Hemoglobin synthesis disorders (e.g., alpha-thalassemias) -RBC membrane abnormalities (e.g., hereditary spherocytosis, elliptocytosis) -Hemangiomas (Kasabach Merritt syndrome) -Acquired conditions, such as sepsis, infections with TORCH or Parvovirus B19 (anemia due to RBC aplasia) and hemolysis secondary to drugs. ISOIMMUNIZATION A. Rh disease (Rh = Rhesus factor) (1) Genetics: Rh positive (+) denotes presence of D antigen. The number of antigenic sites on RBCs varies with genotype. Prevalence of genotype varies with the population. Rh negative (d/d) individuals comprise 15% of Caucasians, 5.5% of African Americans, and <1% of Asians. A sensitized Rh negative mother produces anti-Rh IgG antibodies that cross the placenta. Risk factors for antibody production include 2nd (or later) pregnancies*, maternal toxemia, paternal zygosity (D/D rather than D/d), feto-maternal compatibility in ABO system and antigen load. (2) Clinical presentation of HDN varies from mild jaundice and anemia to hydrops fetalis (with ascites, pleural and pericardial effusions). Because the placenta clears bilirubin, the chief risk to the fetus is anemia. Extramedullary hematopoiesis (due to anemia) results in hepatosplenomegaly. -

Association Between ABO and Duffy Blood Types and Circulating Chemokines and Cytokines
Genes & Immunity (2021) 22:161–171 https://doi.org/10.1038/s41435-021-00137-5 ARTICLE Association between ABO and Duffy blood types and circulating chemokines and cytokines 1 2 3 4 5 6 Sarah C. Van Alsten ● John G. Aversa ● Loredana Santo ● M. Constanza Camargo ● Troy Kemp ● Jia Liu ● 4 7 8 Wen-Yi Huang ● Joshua Sampson ● Charles S. Rabkin Received: 11 February 2021 / Revised: 30 April 2021 / Accepted: 17 May 2021 / Published online: 8 June 2021 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2021, corrected publication 2021 Abstract Blood group antigens are inherited traits that may play a role in immune and inflammatory processes. We investigated associations between blood groups and circulating inflammation-related molecules in 3537 non-Hispanic white participants selected from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Whole-genome scans were used to infer blood types for 12 common antigen systems based on well-characterized single-nucleotide polymorphisms. Serum levels of 96 biomarkers were measured on multiplex fluorescent bead-based panels. We estimated marker associations with blood type using weighted linear or logistic regression models adjusted for age, sex, smoking status, and principal components of p 1234567890();,: 1234567890();,: population substructure. Bonferroni correction was used to control for multiple comparisons, with two-sided values < 0.05 considered statistically significant. Among the 1152 associations tested, 10 were statistically significant. Duffy blood type was associated with levels of CXCL6/GCP2, CXCL5/ENA78, CCL11/EOTAXIN, CXCL1/GRO, CCL2/MCP1, CCL13/ MCP4, and CCL17/TARC, whereas ABO blood type was associated with levels of sVEGFR2, sVEGFR3, and sGP130. -

Blood Product Replacement: Obstetric Hemorrhage
CMQCC OBSTETRIC HEMORRHAGE TOOLKIT Version 2.0 3/24/15 BLOOD PRODUCT REPLACEMENT: OBSTETRIC HEMORRHAGE Richard Lee, MD, Los Angeles County and University of Southern California Medical Center Laurence Shields, MD, Marian Regional Medical Center/Dignity Health Holli Mason, MD, Cedars-Sinai Medical Center Mark Rollins, MD, PhD, University of California, San Francisco Jed Gorlin, MD, Innovative Blood Resources/Memorial Blood Center, St. Paul, Minnesota Maurice Druzin, MD, Lucile Packard Children’s Hospital Stanford University Jennifer McNulty, MD, Long Beach Memorial Medical Center EXECUTIVE SUMMARY • Outcomes are improved with early and aggressive intervention. • Both emergency blood release and massive transfusion protocols should be in place. • In the setting of significant obstetric hemorrhage, resuscitation transfusion should be based on vital signs and blood loss and should not be delayed by waiting for laboratory results. • Calcium replacement will often be necessary with massive transfusion due to the citrate used for anticoagulation in blood products. • During massive transfusion resuscitation, the patient’s arterial blood gas, electrolytes, and core temperature should be monitored to guide clinical management and all transfused fluids should be warmed; direct warming of the patient should be initiated as needed to maintain euthermia and to avoid added coagulopathy. BACKGROUND AND LITERATURE REVIEW After the first several units of packed red blood cells (PRBCs) and in the face of continuing or worsening hemorrhage, aggressive transfusion therapy becomes critical. This report covers the experience with massive transfusion protocols. Lessons from military trauma units as well as civilian experience with motor vehicle accidents and massive obstetric hemorrhage have identified new principles such as earlier use of plasma (FFP/thawed plasma/plasma frozen within 24 hours/liquid plasma) and resuscitation transfusion while laboratory results are pending. -

Fluid Resuscitation Therapy for Hemorrhagic Shock
CLINICAL CARE Fluid Resuscitation Therapy for Hemorrhagic Shock Joseph R. Spaniol vides a review of the 4 types of shock, the 4 classes of Amanda R. Knight, BA hemorrhagic shock, and the latest research on resuscita- tive fluid. The 4 types of shock are categorized into dis- Jessica L. Zebley, MS, RN tributive, obstructive, cardiogenic, and hemorrhagic Dawn Anderson, MS, RN shock. Hemorrhagic shock has been categorized into 4 Janet D. Pierce, DSN, ARNP, CCRN classes, and based on these classes, appropriate treatment can be planned. Crystalloids, colloids, dopamine, and blood products are all considered resuscitative fluid treat- ment options. Each individual case requires various resus- ■ ABSTRACT citative actions with different fluids. Healthcare Hemorrhagic shock is a severe life-threatening emergency professionals who are knowledgeable of the information affecting all organ systems of the body by depriving tissue in this review would be better prepared for patients who of sufficient oxygen and nutrients by decreasing cardiac are admitted with hemorrhagic shock, thus providing output. This article is a short review of the different types optimal care. of shock, followed by information specifically referring to hemorrhagic shock. The American College of Surgeons ■ DISTRIBUTIVE SHOCK categorized shock into 4 classes: (1) distributive; (2) Distributive shock is composed of 3 separate categories obstructive; (3) cardiogenic; and (4) hemorrhagic. based on their clinical outcome. Distributive shock can be Similarly, the classes of hemorrhagic shock are grouped categorized into (1) septic; (2) anaphylactic; and (3) neu- by signs and symptoms, amount of blood loss, and the rogenic shock. type of fluid replacement. This updated review is helpful to trauma nurses in understanding the various clinical Septic shock aspects of shock and the current recommendations for In accordance with the American College of Chest fluid resuscitation therapy following hemorrhagic shock. -

PERSONAL INFORMATION Francesco Rodeghiero WORK
Curriculum Vitae PERSONAL INFORMATION Francesco Rodeghiero WORK EXPERIENCE February 2004- Present Scientific Director Fondazione Progetto Ematologia (Italy) August 2001-October 2015 Director of the Department of Cell Therapy and Hematology Azienda ULSS N.6 (Italy) <p>The Department includes a Unit for Bone Marrow Transplantation, a specialized Center for the diagnosis and treatment of Hemophilia and Thrombosis, and a Research Laboratory</p> 1989- 2016 Professor at the Postgraduate School of Hematology on a contract-basis University of Verona (Italy) February 1993-October 2015 Director of the Hematology Department Azienda ULSS N.6 (Italy) December 1985-October 2015 Director of the Hemostasis and Trombosis Center Azienda ULSS N.6 (Italy) September 2006-November 2016 Member; Chairman from June 2016 to November 2016 Ethics Committee on drugs research and investigational protocol studies of the Vicenza District (Italy) EDUCATION AND TRAINING July 1975- Degree in Medicine University School of Medicine (Italy) December 1984- Postgraduate specialization in Laboratory Medicine University of Padova (Italy) June 1981- Postgraduate specialization in Oncology University of Ferrara (Italy) July 1978- Postgraduate specialization in Hematology University of Ferrara (Italy) ADDITIONAL INFORMATION 15/12/2020 European Medicine Agency Page 1/46 Expertise He has been conducting clinical research in the fields of hematology and hemostasis since the early 1970s. His main interests include thrombocytopenia, hemophilia, von Willebrand disease, thrombophilia, acute promyelocytic leukemia, myeloma, policytemia vera, and the epidemiological aspects of haematological diseases. In the last decades he mainly devoted to clinical research in the field of ITP. Publications 1.T. Barbui, F. Rodeghiero, E. Dini The aspirin tolerance test in von Willebrands disease. -
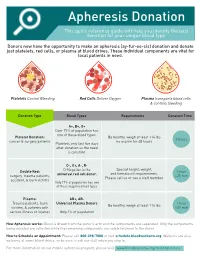
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

Defining Natural Antibodies
PERSPECTIVE published: 26 July 2017 doi: 10.3389/fimmu.2017.00872 Defining Natural Antibodies Nichol E. Holodick1*, Nely Rodríguez-Zhurbenko2 and Ana María Hernández2* 1 Department of Biomedical Sciences, Center for Immunobiology, Western Michigan University Homer Stryker M.D. School of Medicine, Kalamazoo, MI, United States, 2 Natural Antibodies Group, Tumor Immunology Division, Center of Molecular Immunology, Havana, Cuba The traditional definition of natural antibodies (NAbs) states that these antibodies are present prior to the body encountering cognate antigen, providing a first line of defense against infection thereby, allowing time for a specific antibody response to be mounted. The literature has a seemingly common definition of NAbs; however, as our knowledge of antibodies and B cells is refined, re-evaluation of the common definition of NAbs may be required. Defining NAbs becomes important as the function of NAb production is used to define B cell subsets (1) and as these important molecules are shown to play numerous roles in the immune system (Figure 1). Herein, we aim to briefly summarize our current knowledge of NAbs in the context of initiating a discussion within the field of how such an important and multifaceted group of molecules should be defined. Edited by: Keywords: natural antibody, antibodies, natural antibody repertoire, B-1 cells, B cell subsets, B cells Harry W. Schroeder, University of Alabama at Birmingham, United States NATURAL ANTIBODY (NAb) PRODUCING CELLS Reviewed by: Andre M. Vale, Both murine and human NAbs have been discussed in detail since the late 1960s (2, 3); however, Federal University of Rio cells producing NAbs were not identified until 1983 in the murine system (4, 5). -

Blood Product Modifications: Leukofiltration, Irradiation and Washing
Blood Product Modifications: Leukofiltration, Irradiation and Washing 1. Leukocyte Reduction Definitions and Standards: o Process also known as leukoreduction, or leukofiltration o Applicable AABB Standards, 25th ed. Leukocyte-reduced RBCs At least 85% of original RBCs < 5 x 106 WBCs in 95% of units tested . Leukocyte-reduced Platelet Concentrates: At least 5.5 x 1010 platelets in 75% of units tested < 8.3 x 105 WBCs in 95% of units tested pH≥6.2 in at least 90% of units tested . Leukocyte-reduced Apheresis Platelets: At least 3.0 x 1011 platelets in 90% of units tested < 5.0 x 106 WBCs 95% of units tested pH≥6.2 in at least 90% of units tested Methods o Filter: “Fourth-generation” filters remove 99.99% WBCs o Apheresis methods: most apheresis machines have built-in leukoreduction mechanisms o Less efficient methods of reducing WBC content . Washing, deglycerolizing after thawing a frozen unit, centrifugation . These methods do not meet requirement of < 5.0 x 106 WBCs per unit of RBCs/apheresis platelets. Types of leukofiltration/leukoreduction o “Pre-storage” . Done within 24 hours of collection . May use inline filters at time of collection (apheresis) or post collection o “Pre-transfusion” leukoreduction/bedside leukoreduction . Done prior to transfusion . “Bedside” leukoreduction uses gravity-based filters at time of transfusion. Least desirable given variability in practice and absence of proficiency . Alternatively performed by transfusion service prior to issuing Benefits of leukoreduction o Prevention of alloimmunization to donor HLA antigens . Anti-HLA can mediate graft rejection and immune mediated destruction of platelets o Leukoreduced products are indicated for transplant recipients or patients who are likely platelet transfusion dependent o Prevention of febrile non-hemolytic transfusion reactions (FNHTR) . -

45 Part 606—Current Good Man- Ufacturing Practice
Food and Drug Administration, HHS Pt. 606 a presentation. The presiding officer ucts approved under § 601.91, the re- may, as a matter of discretion, permit strictions would no longer apply when questions to be submitted to the pre- FDA determines that safe use of the bi- siding officer for response by a person ological product can be ensured making a presentation. through appropriate labeling. FDA also (f) Judicial review. The Commissioner retains the discretion to remove spe- of Food and Drugs’ decision constitutes cific postapproval requirements upon final agency action from which the ap- review of a petition submitted by the plicant may petition for judicial re- sponsor in accordance with § 10.30 of view. Before requesting an order from a this chapter. court for a stay of action pending re- view, an applicant must first submit a PART 606—CURRENT GOOD MAN- petition for a stay of action under § 10.35 of this chapter. UFACTURING PRACTICE FOR BLOOD AND BLOOD COMPO- [67 FR 37996, May 31, 2002, as amended at 70 NENTS FR 14984, Mar. 24, 2005] § 601.93 Postmarketing safety report- Subpart A—General Provisions ing. Sec. Biological products approved under 606.3 Definitions. this subpart are subject to the post- marketing recordkeeping and safety Subpart B—Organization and Personnel reporting applicable to all approved bi- ological products. 606.20 Personnel. § 601.94 Promotional materials. Subpart C—Plant and Facilities For biological products being consid- 606.40 Facilities. ered for approval under this subpart, unless otherwise informed by the agen- Subpart D—Equipment cy, applicants must submit to the agency for consideration during the 606.60 Equipment. -

Intraoperative Fluid Therapy and Pulmonary Complications
■ Feature Article Intraoperative Fluid Therapy and Pulmonary Complications KRZYSZTOF SIEMIONOW, MD; JACEK CYWINSKI, MD; KRZYSZTOF KUSZA, MD, PHD; ISADOR LIEBERMAN, MD, MBA, FRCSC abstract Full article available online at ORTHOSuperSite.com. Search: 20120123-06 The purpose of this study was to evaluate the effects of intraoperative fl uid therapy on length of hospital stay and pulmonary complications in patients undergoing spine surgery. A total of 1307 patients were analyzed. Sixteen pulmonary complications were observed. Patients with a higher volume of administered crystalloids, colloids, and total intravenous fl uids were more likely to have postoperative respiratory com- plications: the odds of postoperative respiratory complications increased by 30% with an increase of 1000 mL of crystalloid administered. The best cutoff point for total fl uids was 4165 mL, with a sensitivity of 0.8125 and specifi city of 0.7171, for postoperative pulmonary complications. A direct correlation existed between fl uids and length of stay: patients who received Ͼ4165 mL of total fl uids had an average length of stay of 3.88Ϯ4.66 days vs 2.3Ϯ3.9 days for patients who received Ͻ4165 mL of total fl uids (PϽ.0001). This study should be considered as hypothesis-generating to design a prospective trial comparing high vs low intraoperative fl uid regiments for patients undergoing spine surgery. Dr Siemionow is from the Department of Orthopaedic Surgery, University of Illinois, Chicago, Illinois; Dr Cywinski is from the Department of Anesthesia, Cleveland Clinic, Cleveland, Ohio; Dr Kusza is from the Department of Anesthesia, Centrum Medyczne Bydgoszcz, Bydgoszcz, Poland; and Dr Lieberman is from Texas Back Institute, Plano, Texas.