20Th-Century Industrial Black Carbon Emissions Altered Arctic Climate Forcing Joseph R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Read the Article As
Helena Forshell ROCK ART An attempt to understand rock art motifs through ancient literature, epics of creation and the history of metals IN WORKS AND DAYS (probably written down that the different properties of the metals in the 9th century B.C.) by the Greek Hesiod, the may have brought about the growing cruelty first humans are described as a golden race, and greed of man. In which way may the so- ”living like gods without sorrow of heart, re- cial behaviour of man be influenced by the mote and free from toil and grief. When they availability of metals and knowledge of their died, it was as though they were overcome properties? First, a brief outline of the qualities with sleep … they had all good things; for of gold, silver and copper. the fruitful earth unforced bare them fruit abundantly and without stint”. “The second generation which was of silver was less no- Gold ble by far … when they were full grown and Thousands of gold occurrences, most of them were come to the full measure of there prime, less profitable have been discovered in Europe they lived only a little time and that in sorrow alone. There, and in Caucasus, Ural and the because of their foolishness, for they could Himalayas, the heavy gold has accumulated not keep from sinning and from wronging in sea-beds and rivers through erosion of the one another”. The third generation, that of rock. Already in prehistoric times, gold was bronze, was violent, ”… they ate no bread, sifted in large quantities. Parrish (p. -

Ancient Foundations Unit Two CA * the Babylonians
Marshall High School Mr. Cline Western Civilization I: Ancient Foundations Unit Two CA * The Babylonians • By the mid-20th century BC, Sumerian Civilization had already been through a lot: • It had been co opted by the Akkadians • It had been conquered by the Guti. • It had thrown off its invaders, and started a new Sumerian Empire with Ur as its capitol • Then, in 1950 BC, a new group of people entered the scene, the Elamites, a fierce people living to the southeast of Mesopotamia. • The Elamites, like the Guti before them, seem to have been more interested in pillaging than empire building. • It would take another thousand years before the Elamites would mount their own bid for control of the empire. • Nevertheless, the Elamites destroyed the power structure that held the Sumerian empire together. • After a thousand years, the Sumero-Akkadian empire was dead at last. * The Babylonians • Yet the idea of a united Mesopotamian empire lived on as new peoples tried their hand at imperialism. • With the break down of the empire at the hands of the Elamites, a new people, the Amorites, came to conquer much of southern Mesopotamia, including an important religious center called Babylon. • Like the Sumerians before them, the Amorites began by creating minor kingdoms or city states, which vied with one another for power. • The earliest of these were two cities, Isin and Larsa. • For about 200 years, these two were rivals and struggled with each other for supremacy. • Then around 1830, the city of Babylon took advantage of the distraction of these two power players and established itself as an independent kingdom. -

Dating Tablets in the Garshana Corpus
Proceedings of the Thirtieth International Florida Artificial Intelligence Research Society Conference Dating Tablets in the Garshana Corpus James Hearne, Connor Anderson, Yudong Liu Dannon Dixon, Dario Fenstermacher Ritchie Computer Science Department Western Washington University Bellingham, Washington 98226 Abstract original state-owned archives from which they came, depriv- ing scholars of the ability to reconstruct local economic ac- This paper reports on an effort to assign dates to undated tivity. Tablets originally stored in the same local archive are tablets in the Garshana corpus, a fully curated and annotated now scattered throughout the world in museums and univer- collection of Sumerian tables from the Ur III era. Of the 1488 tablets, 92% are dated, giving a strong training set for dat- sities. The ability to identify dense regions of a larger social ing those undated. Two approaches were pursued: (1) a naive network, quasi-cliques, would support the assignment of a one which determined the date of a tablet by simply counting tablet to a specific but now scattered archive. Since names the name overlap with tablets of known year and (2) invok- were reused in Sumerian society, as they are in most, the ing a collection of machine learning algorithms of established identification of tablet dates is essential for such an assign- robustness. The naive method reports an accuracy of 45-55% ment. and the machine learning algorithms achieve 78.8-84.15% ac- This overarching goal requires the identification of two curacy. sorts of information from the original tablets, dates and per- sonal names. It also requires, as this study reveals, the iden- Introduction tification of geographical names. -
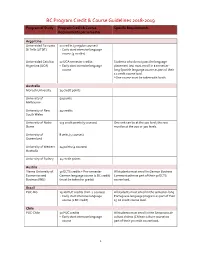
BC Program Credit & Course Guidelines 2018-2019
BC Program Credit & Course Guidelines 2018-2019 Program of Study Program Credit & Course Specific Requirements Requirements per semester Argentina Universidad Torcuato 12 credits (3 regular courses) Di Tella (UTDT) + Early start intensive language course (3 credits) Universidad Catolica 12 UCA semester credits Students who do not pass the language Argentina (UCA) + Early start intensive language placement test must enroll in a semester- course long Spanish language course as part of their 12 credit course load. *One course must be taken with locals Australia Monash University 24 credit points University of 50 points Melbourne University of New 24 credits South Wales University of Notre 125 credit points (5 courses) One unit can be at the 100 level; the rest Dame must be at the 200 or 300 levels. University of 8 units (4 courses) Queensland University of Western 24 points (4 courses) Australia University of Sydney 24 credit points Austria Vienna University of 30 ECTS credits + Pre-semester All students must enroll in German Business Economics and German language course (1 BC credit) Communication as part of their 30 ECTS Business (WU) (must be taken for grade) course load. Brazil PUC-Rio 15-16 PUC credits (min. 4 courses) All students must enroll in the semester-long + Early start intensive language Portuguese language program as part of their course (1 BC credit) 15-16 credit course load. Chile PUC-Chile 50 PUC credits All students must enroll in the Semanario de + Early start intensive language cultura chilena (Chilean culture course) as course part of their 50 credit course load. 1 BC Program Credit & Course Guidelines 2018-2019 Program of Study Program Credit & Course Specific Requirements Requirements per semester Universidad Alberto 50 AH credits All students must enroll in the intensive early Hurtado (AH) start language program as part of their 50 credit course load. -

Human Reproduction and Congenital Heart Disease
Human Reproduction and Congenital Heart Disease: From Biblical/Mythic to Scientific ANCIENT GREECE Pregnancy was regarded as a natural occurrence, not a medical condition. Women in labor were excluded from the sacred sanctuary of Aesclepius because they did not have an ailment that required treatment. Asexual Human Births Birth of Eve--From the rib of Adam Birth of Athena--From the head of Zeus Birth of Aphrodite--From the spume of the sea Virgin Births The Birth of Eve. “And the rib, which the Lord God had taken from man, made he a woman.” Adam and Eve 16th Century Before the Rennaisance, theologians taught that Adam and Eve did not have navels because they were not born of woman. Athena was born of Zeus without a mother. All by himself Zeus fathered grey-eyed Athena. The goddess, fully grown and fully armed, leaped from his head. Botticelli’s Birth of Venus Round the divine flesh rose up a colorless foam whence grew a maiden who was called Aphrodite because she was born from the spume of the sea. Virgin Births The earliest account of virgin births The Pregnant Virgin was from ancient China circa 600 BC. Piero della Francesca, The offspring were always male. 1460 Miraculous birth enhanced the stature of dynastic rulers. 456 BC Aeschylus The mother is not the true parent of the child, but instead serves as a passive repository, a nurse who tends the growth of the seed planted by its true parent-- the father. 2006 In vitro Fertilization The mother is not the true parent of the child but instead serves as a passive repository, a nurse who tends the growth of the implanted seed--the donor eggs. -

Edward Paul Stratford Department of History Brigham Young University Provo, UT 84602 (801) 422 8556 Edward [email protected]
Edward Paul Stratford Department of History Brigham Young University Provo, UT 84602 (801) 422 8556 [email protected] Academic Positions 2010–Present Assistant Professor, Department of History, Brigham Young University, Provo, UT. 2009-2010 Instructor, Department of History, Brigham Young University, Provo, UT. 2007-2009 Research Assistant, Chicago Assyrian Dictionary, Oriental Institute, University of Chicago, Chicago, IL. 2006-2007 Visiting Research Associate, Old Assyrian Text Project, Copenhagen, Denmark. Education Ph.D., 2010 Department of Near Eastern Languages and Civilizations, University of Chicago, Chicago, IL. Defended with Honors. Dissertation: “Archives, Agents, and Risk: An Account of Assyrian Commerce in 1894 B.C.” Publications “Old Assyrian Literacy: Formulating a Method for Graphic Analysis and Some Initial Results” KIM 1: Proceedings of the 1st Kültepe International Meeting, Kültepe 12-23 September, 2013, Subartu 35. ed. Kulakoğlu, Fikri and Cécile Michel (Brepols: Turnhout, Belgium, 2015), p. 117- 128. “Intercalation, Successor Eponyms, and Winter in the Old Assyrian Period: A Critical Reappraisal,” Journal of Near Eastern Studies 74:301-24 (2015). “‘Make Them Pay’: Charting the Social Topography of an Old Assyrian Venture” Journal of Cuneiform Studies 66:11-38 (2014). “Old Assyrian Prices,” in World History Encyclopedia, ed. Alfred J. Andrea (ABC-Clio, 2009). “Old Assyrian Trade Network,” in World History Encyclopedia, ed. Alfred J. Andrea (ABC-Clio, 2009). Works in Progress “Non-destructive provenance study of Old Assyrian cuneiform tablets using portable X-ray flourescence (pXRF)” (in preparation). “Caravans, Cuneiform, and Clay: Beginning a Social Geography of Anatolian Geography during the Old Assyrian period through pXRF Analysis” (in preparation). “Merchant Letters, Human Time” (in preparation). -

Recognizing Proper Names in UR III Texts Through Supervised Learning
Proceedings of the Twenty-Ninth International Florida Artificial Intelligence Research Society Conference Recognizing Proper Names in UR III Texts through Supervised Learning Yudong Liu, James Hearne and Bryan Conrad Computer Science Department Western Washington University Bellingham, Washington 98226 {[email protected]@-bconrad@students.}wwu.edu Abstract names on the tablets suggests the possibility of reconstruct- This paper reports on an ongoing effort to provide computa- ing social networks of actors in the mercantile class and also, tional linguistic support to scholars making use of the writ- given the overlap, their social network connections to roy- ings from the Third Dynasty of Ur, especially those trying alty. Use of the tablets for such purposes is impeded in three to link reports of financial transactions together for the pur- ways. First, they are by and large written in Sumerian, a lan- pose of social networking. The computational experiments guage already going out of use as the language of the streets presented are especially addressed to the problem of identify- at the time the tablets were being written and which, even ing proper names for the ultimate purpose of reconstructing after nearly two centuries of scholarly investigation, is ill- a social network of UR III society. We describe the applica- understood. A casual exploration conducted by us showed tion of established supervised learning algorithms, compare that roughly half of the lexical items found in the UR III Cor- its results to previous work using unsupervised methods and pus are hapax-legomena, words having but a single attesta- propose future work based upon these comparative results. -

Rafał Kolinski 20Th Century BC in the Khabur Triangle Region and The
Rafał Koli´nski 20th Century BC in the Khabur Triangle Region and the Advent of the Old Assyrian Trade with Anatolia 0. Introduction For some time, the turn of the third millennium BC in Northern Mesopotamia has been considered a period of settlement collapse that witnessed the abandonment of the whole area (Weiss et al. 1993). Re- cent research in the Khabur Triangle, a region located in the central part of the Jazirah, has demon- strated that during the so-called Post-Akkadian (or Early Jazirah V) period the central part of the area at least had been settled and probably politically divided between Urkesˇ and Nagar, both serving as seats of local dynasties of Hurrian origin. A dark period starts with the abandonment of the Post-Akkadian le- vels at most of the sites in the area. As for the subsequent period, lasting approximately 100 to 200 years little or nothing is known about the archaeology of the area. Only with the advent of the Middle Bronze Age II period, in which the well-known Khabur Ware pottery appears in its technically and stylistically most developed form, do settlements in the Khabur Triangle seem to be reestablished. Yet, this pro- cess can hardly be dated with precision. At some sites Khabur Ware is contemporary with Samsı¯-Addu’s ‘Kingdom of North Mesopotamia’1 (1809–1776 BC) (Barjamovic et al., 25, 29–32, fig. 10) such as at Tell Chagar Bazar, Tell Rimah, Tell Leilan, and Tell Bi’a, i.e., belong to the first quarter of 18th century BC2, but it appears as well on sites with later texts, dating after the Samsı¯-Addu’s death.3 On the other hand, pottery earlier than the 18th century BC has turned out to be very difficult to identify (Faivre / Nicolle 2007, 185). -

Middle Bronze Age Jerusalem
Radiocarbon, Vol 63, Nr 3, 2021, p 853–883 DOI:10.1017/RDC.2021.21 © The Author(s), 2021. Published by Cambridge University Press for the Arizona Board of Regents on behalf of the University of Arizona. This is an Open Access article, distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike licence (http://creativecommons.org/licenses/ by-nc-sa/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the same Creative Commons licence is included and the original work is properly cited. The written permission of Cambridge University Press must be obtained for commercial re-use. MIDDLE BRONZE AGE JERUSALEM: RECALCULATING ITS CHARACTER AND CHRONOLOGY Johanna Regev1 • Yuval Gadot2 • Helena Roth2 • Joe Uziel3 • Ortal Chalaf3 • Doron Ben-Ami3 • Eugenia Mintz1 • Lior Regev1 • Elisabetta Boaretto1* 1D-REAMS Radiocarbon Laboratory, Scientific Archaeology Unit, Weizmann Institute of Science, Rehovot 7610001, Israel 2The Lester and Sally Entin Faculty of Humanities, Tel Aviv University, Tel-Aviv 6997801, Israel 3Israel Antiquities Authority, PO Box 586, Jerusalem 9100402, Israel ABSTRACT. The following paper presents the results of radiocarbon (14C) dating of Middle Bronze Age (MB) contexts in Jerusalem. The dates, sampled with microarchaeology methods from three different locations along the eastern slopes of the city’s ancient core, reveal that Jerusalem was initially settled in the early phases of the period, with public architecture first appearing in the beginning of the 19th century BC and continued to develop until the 17th century BC. At that time, a curious gap in settlement is noted until the 16th century BC, when the site is resettled. -

Joanna Jędrysik and Marcin S. Przybyła the Early Bronze Age
T e Stone Fortif cations of the Maszkowice Hillfort 141 Joanna Jędrysik and Marcin S. Przybyła T e Early Bronze Age Stone Fortif cations of the Maszkowice Hillfort (Polish Carpathians). Product of an Adaptive Mind or idée f xe? Among many prehistoric hillforts of the Western Carpathians the one located at Maszkowice village dis- plays unique traits. T e site was excavated in 1960s and 1970s, but it was not until 2015 that the new f eld project revealed remains of massive stone fortif cations. T e wall of the Zyndram’s Hill is dated to the Early Bronze Age (18th century BC), being one of the earliest examples of defensive stone architecture in Europe outside Mediterranean. In our paper we shall discuss the development of the defensive system with its geographical and settlement context. Considering the results of f eldwork and other applied methods we can assume, that the enclosed settlement in Maszkowice functioned as an isolated point located in scarcely populated area. T erefore, we need to stress the landscape and geological circumstances which played a signif cant role in inner layout organization, social perception and the development of settlement and its fortif cations. T e stone wall was erected already at the beginning of the site’s occupation. T e defensive system existed then in its most elaborated form (with at least two gates leading into the village), while later during several dozen years the fortif cations slowly but constantly deteriorated. Finally, in conclusion we shall consider the stone wall of Zyndram’s Hill not as a product of local adaptation, but as a result of a pre- pared execution of a project. -
Book of Centuries, They Are for Your Information on How to Use the Book
Mapping History A Timeline Book of the Centuries by Michele Quigley Contents: • Cover page • Introduction • Labeling worksheet sample • Blank labeling worksheet • 2 page spread sample pages - 13th century BC • 2 page spread sample pages - 13th century AD • Full color cover • Bookplate page • 3 blank pages • 48 lined, columned pages • 3 blank pages Printing Instructions: The left side of every page is intended to be blank. Therefore you will simply print this file as is and your book will be complete. After printing you can either 3 hole punch the pages and put them in a binder of take them to a printer to be bound together Please note: The first eight pages --cover page, introduction, labeling worksheets and sample pages-- are not meant to be included in the actual book of centuries, they are for your information on how to use the book. The Book of Centuries begins with the full color cover on page 9. © 2013 Michele Quigley. For Personal Use Only. Students in the PNEU schools would begin a “Book of the Centuries” around the age of 10 and keep the book throughout their school years. A child should have a concept of time and the past before beginning his centuries book. If you wish to begin earlier, you might consider making a “family” book of centuries. Each two page spread represents 100 years of history. The left side is blank, the right side is lined and columned. (see sample pages) On the right the child will record historical events, names and dates on and on the left he will make illustrations of artifacts, tools, pottery, clothing, etc., of the time period. -

On the Theory of Demographic Cycles
Translation of the 2003 paper published in Russian, academic journal Vostok-Oriens 3:5-22. * Sergey Nefedov The theory of demographic (or ecological) cycles is one of the new directions in economic and social history; the distinctive feature of this direction is application of population dynamic models. American biologist and demographer Raymond Pearl laid the foundation of the theory of demographic cycles by showing that the quantitative changes of animal (and perhaps human) populations may be described by the so-called logistic equation (Pearl 1926). The solution of this differential equation is the logistic curve (fig. 1). The course of the logistic curve shows that at first, when resources are abundant and consumption level is high population grows rapidly. However, because resources are limited, population growth results in a decline of consumption per capita (curve 2 on fig. 1). We can observe such situation, for example, in China of the Qing dynasty (Fig.2. Consumption level is measured by the amount of liters of rice that an unskilled worker could buy with his day salary1.) The decrease of consumption causes a deceleration in population growth, which approaches an asymptote corresponding to the maximum possible number of the population when all the natural resources are used up. Achieving the maximum possible level means that people subsist at the minimal consumption level, practically on the verge of extinction, so that the natural growth is completely eliminated. This state of “hungry homeostasis” is in fact unstable. Fluctuations of natural factors can lead to “demographic crash”, disastrous starvation, or an epidemic. Such a disaster results to a sudden population decrease, followed by the period of reconstruction in a new demographic cycle.