Chapter 11 – Coordination Chemistry: Bonding, Spectra, and Magnetism Read Through the Top of P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Powerful Framework for the Chemical Design of Molecular Nanomagnets Yan Duan†,1,2, Joana T
Data mining, dashboards and statistics: a powerful framework for the chemical design of molecular nanomagnets Yan Duan†,1,2, Joana T. Coutinho†,*,1,3, Lorena E. Rosaleny†,*,1, Salvador Cardona-Serra1, José J. Baldoví1, Alejandro Gaita-Ariño*,1 1 Instituto de Ciencia Molecular (ICMol), Universitat de València, C/ Catedrático José Beltrán 2, 46980 Paterna, Spain 2 Department of Chemistry and the Ilse Katz Institute for Nanoscale Science & Technology, Ben-Gurion University of the Negev, Beer Sheva 84105, Israel 3 Centre for Rapid and Sustainable Product Development, Polytechnic of Leiria, 2430-028 Marinha Grande, Portugal *e-mail: [email protected], [email protected], [email protected] Abstract Three decades of intensive research in molecular nanomagnets have brought the magnetic memory in molecules from liquid helium to liquid nitrogen temperature. The enhancement of this operational temperature relies on a wise choice of the magnetic ion and the coordination environment. However, serendipity, oversimplified theories and chemical intuition have played the main role. In order to establish a powerful framework for statistically driven chemical design, we collected chemical and physical data for lanthanide-based nanomagnets to create a catalogue of over 1400 published experiments, developed an interactive dashboard (SIMDAVIS) to visualise the dataset, and applied inferential statistical analysis to it. We found that the effective energy barrier derived from the Arrhenius equation displays an excellent correlation with the magnetic memory, and that among all chemical families studied, only terbium bis-phthalocyaninato sandwiches and dysprosium metallocenes consistently present magnetic memory up to high temperature, but that there are some promising strategies for improvement. -

An Earlier Collection of These Notes in One PDF File
UNIVERSITY OF THE WEST INDIES MONA, JAMAICA Chemistry of the First Row Transition Metal Complexes C21J Inorganic Chemistry http://wwwchem.uwimona.edu.jm/courses/index.html Prof. R. J. Lancashire September 2005 Chemistry of the First Row Transition Metal Complexes. C21J Inorganic Chemistry 24 Lectures 2005/2006 1. Review of Crystal Field Theory. Crystal Field Stabilisation Energies: origin and effects on structures and thermodynamic properties. Introduction to Absorption Spectroscopy and Magnetism. The d1 case. Ligand Field Theory and evidence for the interaction of ligand orbitals with metal orbitals. 2. Spectroscopic properties of first row transition metal complexes. a) Electronic states of partly filled quantum levels. l, ml and s quantum numbers. Selection rules for electronic transitions. b) Splitting of the free ion energy levels in Octahedral and Tetrahedral complexes. Orgel and Tanabe-Sugano diagrams. c) Spectra of aquated metal ions. Factors affecting positions, intensities and shapes of absorption bands. 3. Magnetic Susceptibilities of first row transition metal complexes. a) Effect of orbital contributions arising from ground and excited states. b) Deviation from the spin-only approximation. c) Experimental determination of magnetic moments. Interpretation of data. 4. General properties (physical and chemical) of the 3d transition metals as a consequence of their electronic configuration. Periodic trends in stabilities of common oxidation states. Contrast between first-row elements and their heavier congeners. 5. A survey of the chemistry of some of the elements Ti....Cu, which will include the following topics: a) Occurrence, extraction, biological significance, reactions and uses b) Redox reactions, effects of pH on the simple aqua ions c) Simple oxides, halides and other simple binary compounds. -

Crystal Field Theory (CFT)
Crystal Field Theory (CFT) The bonding of transition metal complexes can be explained by two approaches: crystal field theory and molecular orbital theory. Molecular orbital theory takes a covalent approach, and considers the overlap of d-orbitals with orbitals on the ligands to form molecular orbitals; this is not covered on this site. Crystal field theory takes the ionic approach and considers the ligands as point charges around a central metal positive ion, ignoring any covalent interactions. The negative charge on the ligands is repelled by electrons in the d-orbitals of the metal. The orientation of the d orbitals with respect to the ligands around the central metal ion is important, and can be used to explain why the five d-orbitals are not degenerate (= at the same energy). Whether the d orbitals point along or in between the cartesian axes determines how the orbitals are split into groups of different energies. Why is it required? The valence bond approach could not explain the Electronic spectra, Magnetic moments, Reaction mechanisms of the complexes. Assumptions of CFT: 1. The central Metal cation is surrounded by ligand which contain one or more lone pair of electrons. 2. The ionic ligand (F-, Cl- etc.) are regarded as point charges and neutral molecules (H2O, NH3 etc.) as point dipoles. 3. The electrons of ligand does not enter metal orbital. Thus there is no orbital overlap takes place. 4. The bonding between metal and ligand is purely electrostatic i.e. only ionic interaction. The approach taken uses classical potential energy equations that take into account the attractive and repulsive interactions between charged particles (that is, Coulomb's Law interactions). -

Computational Studies of Three Chemical Systems
Computational Studies of Three Chemical Systems _______________________________________ A Dissertation Presented to The Faculty of the Graduate School University of Missouri-Columbia _______________________________________________________ In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy _____________________________________________________ by Haunani Thomas Prof. Carol A. Deakyne, Dissertation Supervisor December 2011 The undersigned, appointed by the dean of the Graduate School, have examined the dissertation entitled COMPUTATIONAL STUDIES OF THREE CHEMICAL SYSTEMS Presented by Haunani Thomas, a candidate for the degree of doctor of philosophy of Chemistry, and hereby certify that, in their opinion, it is worthy of acceptance. Professor Carol Deakyne (Chair) Professor John Adams (Member) Professor Michael Greenlief (Member) Professor Giovanni Vignale (Outside Member) ACKNOWLEDGEMENTS I am forever indebted to my dissertation supervisor, Professor Carol Deakyne, for her guidance and support. She has been an excellent and patient mentor through the graduate school process, always mindful of the practical necessities of my progress, introducing me to the field of Chemistry, and allowing me to advance professionally through attendance and presentations at ACS meetings. I am fully aware that, in this respect, not all graduate students are as fortunate as I have been. I am also grateful to Professor John Adams for all his support and teaching throughout my graduate career. I am appreciative of my entire committee, Professor Deakyne, Professor Adams, Professor Michael Greenlief, and Professor Giovanni Vignale, for their sound advice, patience, and flexibility. I would also like to thank my experimental collaborators and their groups, Professor Joel Liebman of the University of Maryland, Baltimore County, Professor Michael Van Stip Donk of Wichita State University, and Professor Jerry Atwood of the University of Missouri – Columbia. -

Werner-Type Complexes of Uranium(III) and (IV) Judith Riedhammer, J
pubs.acs.org/IC Article Werner-Type Complexes of Uranium(III) and (IV) Judith Riedhammer, J. Rolando Aguilar-Calderon,́ Matthias Miehlich, Dominik P. Halter, Dominik Munz, Frank W. Heinemann, Skye Fortier, Karsten Meyer,* and Daniel J. Mindiola* Cite This: Inorg. Chem. 2020, 59, 2443−2449 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: Transmetalation of the β-diketiminate salt [M]- Me Ph + Me Ph− − [ nacnac ](M =NaorK; nacnac = {PhNC(CH3)}2CH ) with UI3(THF)4 resulted in the formation of the homoleptic, Me Ph octahedral complex [U( nacnac )3](1). Green colored 1 was fully characterized by a solid-state X-ray diffraction analysis and a combination of UV/vis/NIR, NMR, and EPR spectroscopic studies as well as solid-state SQUID magnetization studies and density functional theory calculations. Electrochemical studies of 1 revealed this species to possess two anodic waves for the U(III/IV) and U(IV/V) redox couples, with the former being chemically accessible. Using mild oxidants, such as [CoCp2][PF6]or [FeCp ][Al{OC(CF ) } ], yields the discrete salts [1][A] (A = − 2 3 −3 4 PF6 , Al{OC(CF3)3}4 ), whereas the anion exchange of [1][PF6] with NaBPh4 yields [1][BPh4]. Me Dipp μ 12 ■ INTRODUCTION UCl3(THF)] and [{( nacnac )UCl}2( 2-Cl)3]Cl. In Me Dipp− ’ some cases, disubstitution of nacnac generates the Alfred Werner s disapproval of the complex ion-chain theory Me Dipp η3 Me Dipp 13 proposed by Jørgensen and Blomstrand,1 via the introduction rearranged species [( nacnac )( - nacnac )UI], of coordination complexes and the concept of Nebenvalenz,1,2 where one ligand has changed hapticity, most likely due to fi steric constraints. -

M.Sc. (Chemistry) M
Regulations 2019 Curriculum and Syllabi (Amendments updated upto June 2020) M.Sc. (Chemistry) M. Sc. Chemistry Regulations 2019 REGULATIONS 2019 CURRICULUM AND SYLLABI (Amendments updated upto June 2020) M.Sc. CHEMISTRY B.S. Abdur Rahman Crescent Institute of Science and Technology 1 M. Sc. Chemistry Regulations 2019 B.S. Abdur Rahman Crescent Institute of Science and Technology 2 M. Sc. Chemistry Regulations 2019 VISION AND MISSION OF THE INSTITUTION VISION B.S.Abdur Rahman Crescent Institute of Science and Technology aspires to be a leader in Education, Training and Research in multidisciplinary areas of importance and to play a vital role in the Socio-Economic progress of the Country in a sustainable manner. MISSION To blossom into an internationally renowned Institute. To empower the youth through quality and value-based education. To promote professional leadership and entrepreneurship. To achieve excellence in all its endeavors to face global challenges. To provide excellent teaching and research ambience. To network with global Institutions of Excellence, Business, Industry and Research Organizations. To contribute to the knowledge base through Scientific enquiry, Applied Research and Innovation. B.S. Abdur Rahman Crescent Institute of Science and Technology 3 M. Sc. Chemistry Regulations 2019 B.S. Abdur Rahman Crescent Institute of Science and Technology 4 M. Sc. Chemistry Regulations 2019 DEPARTMENT OF CHEMISTRY VISION AND MISSION VISION To blossom as a department with excellence in the field of Chemical Sciences through academic and research programmes in cutting-edge areas. MISSION To provide knowledge and skill in Chemical Sciences through post graduate and doctoral programmes. To undertake research in emerging areas of Chemical Sciences and transform the findings for the benefit of the society. -

A New Way for Probing Bond Strength J
A New Way for Probing Bond Strength J. Klein, H. Khartabil, J.C. Boisson, J. Contreras-Garcia, Jean-Philip Piquemal, E. Henon To cite this version: J. Klein, H. Khartabil, J.C. Boisson, J. Contreras-Garcia, Jean-Philip Piquemal, et al.. A New Way for Probing Bond Strength. Journal of Physical Chemistry A, American Chemical Society, 2020, 124 (9), pp.1850-1860. 10.1021/acs.jpca.9b09845. hal-02377737 HAL Id: hal-02377737 https://hal.archives-ouvertes.fr/hal-02377737 Submitted on 27 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. A New Way for Probing Bond Strength Johanna Klein,y Hassan Khartabil,y Jean-Charles Boisson,z Julia Contreras-Garc´ıa,{ Jean-Philip Piquemal,{ and Eric H´enon∗,y yInstitut de Chimie Mol´eculaire de Reims UMR CNRS 7312, Universit´ede Reims Champagne-Ardenne, Moulin de la Housse 51687 Reims Cedex 02 BP39 (France) zCReSTIC EA 3804, Universit´ede Reims Champagne-Ardenne, Moulin de la Housse 51687 Reims Cedex 02 BP39 (France) {Sorbonne Universit´es,UPMC, Laboratoire de Chimie Th´eoriqueand UMR CNRS 7616, 4 Pl Jussieu, 75252 Paris Cedex 05(France) E-mail: [email protected] Phone: +33(3)26918497 1 Abstract The covalent chemical bond is intimately linked to electron sharing between atoms. -

Limitation of Crystal Field Theory the Main Drawback of the Crystal Field Theory Is That It Does Not Consider the Covalent Character in Metal-Ligand Bonding at All
CHAPTER 7 Metal-Ligand Bonding: Limitation of Crystal Field Theory The main drawback of the crystal field theory is that it does not consider the covalent character in metal-ligand bonding at all. It treats the metal-ligand interaction in a purely electrostatic framework which is pretty far from reality. All the effects which originate from covalence cannot be explained by this theory. Therefore, the main limitations of crystal field theory can be concluded only after knowing the causes and magnitude of the covalence in the metal-ligand bonds. Evidences for the Covalent Character in Metal–Ligand Bond The crystal field theory considers the metal center as well as surrounding ligands as point charges and assumes that the interaction between them is 100% ionic. However, quite strong experimental evidences have proved that there is some covalent character too which cannot be ignored. Some of those experimental evidences are as follows: 1. The nephelauxetic effect: The electrons present in the partially filled d-orbitals of the metal center repel each other to produce a number of energy levels. The placement of these levels on the energy scale depends upon the arrangement of filled electrons. The energy of these levels can be given in terms of “Racah parameters” B and C (a measure of interelectronic repulsion). The energy difference between same multiplicity states is expressed in B and Dq while between different multiplicity states is given in term of B, Dq and C. It has been observed that the complexation of metal center always results in a decrease in interelectronic repulsion parameters which in turn also advocates a decrease in the repulsion between d-electron density. -

Searching Coordination Compounds
CAS ONLINEB Available on STN Internationalm The Scientific & Technical Information Network SEARCHING COORDINATION COMPOUNDS December 1986 Chemical Abstracts Service A Division of the American Chemical Society 2540 Olentangy River Road P.O. Box 3012 Columbus, OH 43210 Copyright O 1986 American Chemical Society Quoting or copying of material from this publication for educational purposes is encouraged. providing acknowledgment is made of the source of such material. SEARCHING COORDINATION COMPOUNDS prepared by Adrienne W. Kozlowski Professor of Chemistry Central Connecticut State University while on sabbatical leave as a Visiting Educator, Chemical Abstracts Service Table of Contents Topic PKEFACE ............................s.~........................ 1 CHAPTER 1: INTRODUCTION TO SEARCHING IN CAS ONLINE ............... 1 What is Substructure Searching? ............................... 1 The Basic Commands .............................................. 2 CHAPTEK 2: INTKOOUCTION TO COORDINATION COPPOUNDS ................ 5 Definitions and Terminology ..................................... 5 Ligand Characteristics.......................................... 6 Metal Characteristics .................................... ... 8 CHAPTEK 3: STKUCTUKING AND REGISTKATION POLICIES FOR COORDINATION COMPOUNDS .............................................11 Policies for Structuring Coordination Compounds ................. Ligands .................................................... Ligand Structures........................................... Metal-Ligand -

Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, and a FEW CASE STUDIES
Safety Moment TYLER LAB GROUP MEETING 1 Safety Moment TYLER LAB GROUP MEETING 2 Metal Hydrides: Benchtop vs. Box Hydride = :H- Hydrides are powerful Lewis bases and reducing agent ◦ Exothermically form H2 (this should scare you) ◦ Heating leads to faster reactivity ◦ H evolution leads to rapid increase in pressure2 ◦ Uncontrolled reactions easily cause runaway exotherm, class D fire, explosion, and death/unemployment LiAlH is the #1 chemical cause of fatality in chemical4 industry 3 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” LiAlH4 ◦ Insanely irritating (serious safety hazard) ◦ Extremely moisture sensitive (don’t leave out for >2 minutes) ◦ Ethereal mixtures are pyrophoric! DiBuAl-H ◦ Pyrophoric – it will explode upon exposure to oxygen NaEt3BH ◦ Pyrophoric in solution LiH and NaH ◦ Can be handled on the benchtop (not >2 minutes) ◦ Parrafin oil dispersions much safer KH ◦ Pyrophoric if not in a dispersion ◦ Handle with extreme care! † Sirius Black, Harry Potter and the Prisoner of Azkaban 4 Metal Hydrides: “I want to commit the murder I was imprisoned for†.” CaH2 ◦ Very safe to handle on the benchtop Pt-H, Pd-H, Ni-H ◦ All very pyrophoric NaBH4 ◦ Very safe in general Other hydrides ◦ Treat as pyrophoric ◦ Transition metal hydrides vary in hydridic strength ◦ General rule of thumb: if it does hydrogenations, it is probably pyrophoric ◦ If they’re in organics of any kind, they are probably pyrophoric † Sirius Black, Harry Potter and the Prisoner of Azkaban 5 Organometallic Chemistry BASIC PRINCIPLES, APPLICATIONS, -

Coordination Chemistry (See Chapter 20, H&S 3Rd Ed.)
Coordination Chemistry (see Chapter 20, H&S 3rd Ed.) Transition metal elements, particularly ions, have a strong tendency to form compounds with Lewis bases. Why? Lewis base: electron pair donor egs. H2O, NH3 n+ Lewis acid: electron pair acceptor egs. M , BH3 ‘low lying’ empty orbitals: lower in energy than HOMO of Lewis case Lewis acidity enhanced by positive charge so TM ions are good Lewis bases 2+ 3+ familiar examples: AlCl3, BF3 from main group and M , M in TM series Type of bonding in these compounds? ‘coordinative’ or ‘dative’ bonds to form Lewis base ‘adducts’ L→M TM-Lewis base ‘adducts’ are often referred to as (i) TM ‘complexes’ OR (ii) coordination complexes NB: still polar covalent bonds – this just defines where the pair of electrons came from Some more definitions: ligand: molecule or ion that can act as a Lewis base to a metal - egs. Cl , PR3, ROR (R = generic term for an alkyl) donor atoms: atoms that are in direct contact with the metal ion O in R2O→M denticity: number of donor atoms per ligand; range from 1 to >6 unidentate bidentate tridentate O H O H2N NH2 2 O O water en diglyme N O O O N N O O N porphin 15-C-5 tetradentate pentadentate chelate: multidentate ligands forms rings; M tend to be very stable, OO especially 5 and 6 member chelate rings acac ‘bite’ or ‘bite angle’: a measure of the span or size of a chelate ligand; depends on M-L distance M OO acac coordination complex MLn generic terms commonly used: L neutral Lewis base (electron pair donor) X anionic donor (eg. -
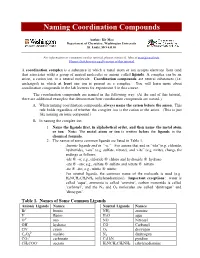
Naming Coordination Compounds
Naming Coordination Compounds Author: Kit Mao Department of Chemistry, Washington University St. Louis, MO 63130 For information or comments on this tutorial, please contact K. Mao at [email protected]. Please click here for a pdf version of this tutorial. A coordination complex is a substance in which a metal atom or ion accepts electrons from (and thus associates with) a group of neutral molecules or anions called ligands. A complex can be an anion, a cation ion, or a neutral molecule. Coordination compounds are neutral substances (i.e. uncharged) in which at least one ion is present as a complex. You will learn more about coordination compounds in the lab lectures for experiment 5 in this course. The coordination compounds are named in the following way. (At the end of this tutorial, there are additional examples that demonstrate how coordination compounds are named.) A. When naming coordination compounds, always name the cation before the anion. This rule holds regardless of whether the complex ion is the cation or the anion. (This is just like naming an ionic compound.) B. In naming the complex ion: 1. Name the ligands first, in alphabetical order, and then name the metal atom or ion. Note: The metal atom or ion is written before the ligands in the chemical formula. 2. The names of some common ligands are listed in Table 1. · Anionic ligands end in “-o.” For anions that end in “-ide”(e.g. chloride, hydroxide), “-ate” (e.g. sulfate, nitrate), and “-ite” (e.g. nirite), change the endings as follows: -ide ® -o; e.g., chloride ® chloro and hydroxide ® hydroxo -ate ® -ato; e.g., sulfate ® sulfato and nitrate ® nitrato -ite ® -ito; e.g., nitrite ® nitrito · For neutral ligands, the common name of the molecule is used (e.g.