Correction for Zhou Et Al., Scaffold Association Factor B (SAFB) Is Required for Expression of Prenyltransferases and RAS Membra
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alterations of Genetic Variants and Transcriptomic Features of Response to Tamoxifen in the Breast Cancer Cell Line
Alterations of Genetic Variants and Transcriptomic Features of Response to Tamoxifen in the Breast Cancer Cell Line Mahnaz Nezamivand-Chegini Shiraz University Hamed Kharrati-Koopaee Shiraz University https://orcid.org/0000-0003-2345-6919 seyed taghi Heydari ( [email protected] ) Shiraz University of Medical Sciences https://orcid.org/0000-0001-7711-1137 Hasan Giahi Shiraz University Ali Dehshahri Shiraz University of Medical Sciences Mehdi Dianatpour Shiraz University of Medical Sciences Kamran Bagheri Lankarani Shiraz University of Medical Sciences Research Keywords: Tamoxifen, breast cancer, genetic variants, RNA-seq. Posted Date: August 17th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-783422/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/33 Abstract Background Breast cancer is one of the most important causes of mortality in the world, and Tamoxifen therapy is known as a medication strategy for estrogen receptor-positive breast cancer. In current study, two hypotheses of Tamoxifen consumption in breast cancer cell line (MCF7) were investigated. First, the effect of Tamoxifen on genes expression prole at transcriptome level was evaluated between the control and treated samples. Second, due to the fact that Tamoxifen is known as a mutagenic factor, there may be an association between the alterations of genetic variants and Tamoxifen treatment, which can impact on the drug response. Methods In current study, the whole-transcriptome (RNA-seq) dataset of four investigations (19 samples) were derived from European Bioinformatics Institute (EBI). At transcriptome level, the effect of Tamoxifen was investigated on gene expression prole between control and treatment samples. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

The Increasing Diversity of Functions Attributed to the SAFB Family of RNA-/DNA-Binding Proteins
Biochemical Journal (2016) 473 4271–4288 DOI: 10.1042/BCJ20160649 Review Article The increasing diversity of functions attributed to the SAFB family of RNA-/DNA-binding proteins Michael Norman1, Caroline Rivers1, Youn-Bok Lee2, Jalilah Idris1 and James Uney1 1Regenerative Medicine Laboratories, School of Clinical Sciences, Cellular and Molecular Medicine, University of Bristol, Medical Sciences Building, University Walk, Bristol BS8 1TD, U.K. and 2Maurice Wohl Clinical Neuroscience Institute, Kings College, 125 Coldharbour Lane, Camberwell, London SE5 9NU, U.K. Correspondence: Michael Norman ([email protected]) RNA-binding proteins play a central role in cellular metabolism by orchestrating the complex interactions of coding, structural and regulatory RNA species. The SAFB (scaf- fold attachment factor B) proteins (SAFB1, SAFB2 and SAFB-like transcriptional modula- tor, SLTM), which are highly conserved evolutionarily, were first identified on the basis of their ability to bind scaffold attachment region DNA elements, but attention has subse- quently shifted to their RNA-binding and protein–protein interactions. Initial studies identi- fied the involvement of these proteins in the cellular stress response and other aspects of gene regulation. More recently, the multifunctional capabilities of SAFB proteins have shown that they play crucial roles in DNA repair, processing of mRNA and regulatory RNA, as well as in interaction with chromatin-modifying complexes. With the advent of new techniques for identifying RNA-binding sites, enumeration of individual RNA targets has now begun. This review aims to summarise what is currently known about the func- tions of SAFB proteins. Introduction In this review, we discuss three scaffold attachment factor B (SAFB) proteins [SAFB1, SAFB2 and SAFB-like transcriptional modulator, SLTM] that bind both DNA and RNA. -

Human Proteins That Interact with RNA/DNA Hybrids
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Resource Human proteins that interact with RNA/DNA hybrids Isabel X. Wang,1,2 Christopher Grunseich,3 Jennifer Fox,1,2 Joshua Burdick,1,2 Zhengwei Zhu,2,4 Niema Ravazian,1 Markus Hafner,5 and Vivian G. Cheung1,2,4 1Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA; 2Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, USA; 3Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, Maryland 20892, USA; 4Department of Pediatrics, University of Michigan, Ann Arbor, Michigan 48109, USA; 5Laboratory of Muscle Stem Cells and Gene Regulation, National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Maryland 20892, USA RNA/DNA hybrids form when RNA hybridizes with its template DNA generating a three-stranded structure known as the R-loop. Knowledge of how they form and resolve, as well as their functional roles, is limited. Here, by pull-down assays followed by mass spectrometry, we identified 803 proteins that bind to RNA/DNA hybrids. Because these proteins were identified using in vitro assays, we confirmed that they bind to R-loops in vivo. They include proteins that are involved in a variety of functions, including most steps of RNA processing. The proteins are enriched for K homology (KH) and helicase domains. Among them, more than 300 proteins preferred binding to hybrids than double-stranded DNA. These proteins serve as starting points for mechanistic studies to elucidate what RNA/DNA hybrids regulate and how they are regulated. -

Interplay of RNA-Binding Proteins and Micrornas in Neurodegenerative Diseases
International Journal of Molecular Sciences Review Interplay of RNA-Binding Proteins and microRNAs in Neurodegenerative Diseases Chisato Kinoshita 1,* , Noriko Kubota 1,2 and Koji Aoyama 1,* 1 Department of Pharmacology, Teikyo University School of Medicine, 2-11-1 Kaga, Itabashi, Tokyo 173-8605, Japan; [email protected] 2 Teikyo University Support Center for Women Physicians and Researchers, 2-11-1 Kaga, Itabashi, Tokyo 173-8605, Japan * Correspondence: [email protected] (C.K.); [email protected] (K.A.); Tel.: +81-3-3964-3794 (C.K.); +81-3-3964-3793 (K.A.) Abstract: The number of patients with neurodegenerative diseases (NDs) is increasing, along with the growing number of older adults. This escalation threatens to create a medical and social crisis. NDs include a large spectrum of heterogeneous and multifactorial pathologies, such as amyotrophic lateral sclerosis, frontotemporal dementia, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and multiple system atrophy, and the formation of inclusion bodies resulting from protein misfolding and aggregation is a hallmark of these disorders. The proteinaceous components of the pathological inclusions include several RNA-binding proteins (RBPs), which play important roles in splicing, stability, transcription and translation. In addition, RBPs were shown to play a critical role in regulating miRNA biogenesis and metabolism. The dysfunction of both RBPs and miRNAs is Citation: Kinoshita, C.; Kubota, N.; often observed in several NDs. Thus, the data about the interplay among RBPs and miRNAs and Aoyama, K. Interplay of RNA-Binding Proteins and their cooperation in brain functions would be important to know for better understanding NDs and microRNAs in Neurodegenerative the development of effective therapeutics. -

Defining the Regulation and Function of SAFB1 and SAFB2 in Human Breast Cancer Cells
Defining the regulation and function of SAFB1 and SAFB2 in human breast cancer cells Elaine Hong Institute of Cellular Medicine Newcastle University A thesis submitted for the degree of Doctor of Philosophy July 2013 Abstract Scaffold attachment factor B1 (SAFB1) and SAFB2 are oestrogen receptor (ER) corepressors that bind and modulate ER activity through chromatin remodelling or interaction with the basal transcription machinery. However, little is known about the fundamental characteristics and function of SAFB1 and SAFB2 proteins in breast cancer. In this study, an investigation of the characteristics and function(s) of SAFB1 and SAFB2 was undertaken; their expression profile was first assessed in ER-positive (MCF-7) and ER-negative (MDA-MB-231) breast cancer cell lines. Results show that SAFB1 and SAFB2 are themselves regulated by an active metabolite of oestrogen, 17β-oestradiol, in both ER positive and ER negative breast cancer cells. Using a combined approach of RNA interference and gene expression profile studies, 12 novel targets closely linked to tumour progression were identified for SAFB1 and SAFB2. Expression levels of the following genes, CDKN2A, CLU, ESR1, IGFBP2, IL2RA, ITGB4, KIT, KLK5, MT3, NGFR and SPRR1B increased while IL-6 expression and secretion decreased when cells were depleted of SAFB proteins. This observation supports their primary role as transcriptional repressors with SAFB2 playing a prominent role in transcriptional regulation in MDA-MB-231 cells. This study has also established a novel link between SAFB proteins and ITGB4 and IL-6 expression. Both SAFB proteins have an internal RNA-recognition motif but little is known about the RNA-binding properties of SAFB1 or SAFB2. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -
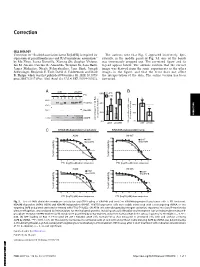
Correction for Zhou Et Al., Scaffold Association Factor B (SAFB) Is Required for Expression of Prenyltransferases and RAS Membra
Correction CELL BIOLOGY Correction for “Scaffold association factor B (SAFB) is required for The authors note that Fig. 5 appeared incorrectly. Spe- expression of prenyltransferases and RAS membrane association,” cifically, in the middle panel of Fig. 5A, one of the bands by Mo Zhou, Leena Kuruvilla, Xiarong Shi, Stephen Viviano, was erroneously cropped out. The corrected figure and its Ian M. Ahearn, Caroline R. Amendola, Wenjuan Su, Sana Badri, legend appear below. The authors confirm that the correct James Mahaffey, Nicole Fehrenbacher, Jane Skok, Joseph image was derived from the same experiments as the other Schlessinger, Benjamin E. Turk, David A. Calderwood, and Mark images in the figure, and that the error does not affect R. Philips, which was first published November 30, 2020; 10.1073/ the interpretation of the data. The online version has been pnas.2005712117 (Proc. Natl. Acad. Sci. U.S.A. 117,31914–31922). corrected. A shNT shSAFB * B 60% - + - + FTI S P S P S P S P Total Ras 40% 100% ** RhoGDI 20% 50% CORRECTION CIMPR in cytosolic fraction FTI treated vs untreated vs untreated FTI treated percentage of total Ras percentage 0% 0% shNT shSAFB GTP-loaded Ras in A549 stables, shNT shSAFB C KRAS4B-dependent lung tumor KRAS4B-independent lung A549 H1437 %cell viability %cell viability FTI (log10, µM) dose response FTI (log10, µM) dose response H358 H1975 %cell viability %cell viability FTI (log10, µM) dose response FTI (log10, µM) dose response Fig. 5. Loss of SAFB diminishes membrane association and GTP loading of KRAS4B and sensitizes KRAS4B-dependent lung tumor cells to FTI treatment. -

The Neurodegenerative Diseases ALS and SMA Are Linked at The
Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gky1093 The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gky1093/5162471 by [email protected] on 06 November 2018 Binkai Chi, Jeremy D. O’Connell, Alexander D. Iocolano, Jordan A. Coady, Yong Yu, Jaya Gangopadhyay, Steven P. Gygi and Robin Reed* Department of Cell Biology, Harvard Medical School, 240 Longwood Ave. Boston MA 02115, USA Received July 17, 2018; Revised October 16, 2018; Editorial Decision October 18, 2018; Accepted October 19, 2018 ABSTRACT Fused in Sarcoma (FUS) and TAR DNA Binding Protein (TARDBP) (9–13). FUS is one of the three members of Understanding the molecular pathways disrupted in the structurally related FET (FUS, EWSR1 and TAF15) motor neuron diseases is urgently needed. Here, we family of RNA/DNA binding proteins (14). In addition to employed CRISPR knockout (KO) to investigate the the RNA/DNA binding domains, the FET proteins also functions of four ALS-causative RNA/DNA binding contain low-complexity domains, and these domains are proteins (FUS, EWSR1, TAF15 and MATR3) within the thought to be involved in ALS pathogenesis (5,15). In light RNAP II/U1 snRNP machinery. We found that each of of the discovery that mutations in FUS are ALS-causative, these structurally related proteins has distinct roles several groups carried out studies to determine whether the with FUS KO resulting in loss of U1 snRNP and the other two members of the FET family, TATA-Box Bind- SMN complex, EWSR1 KO causing dissociation of ing Protein Associated Factor 15 (TAF15) and EWS RNA the tRNA ligase complex, and TAF15 KO resulting in Binding Protein 1 (EWSR1), have a role in ALS.