Correction for Zhou Et Al., Scaffold Association Factor B (SAFB) Is Required for Expression of Prenyltransferases and RAS Membra
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alterations of Genetic Variants and Transcriptomic Features of Response to Tamoxifen in the Breast Cancer Cell Line
Alterations of Genetic Variants and Transcriptomic Features of Response to Tamoxifen in the Breast Cancer Cell Line Mahnaz Nezamivand-Chegini Shiraz University Hamed Kharrati-Koopaee Shiraz University https://orcid.org/0000-0003-2345-6919 seyed taghi Heydari ( [email protected] ) Shiraz University of Medical Sciences https://orcid.org/0000-0001-7711-1137 Hasan Giahi Shiraz University Ali Dehshahri Shiraz University of Medical Sciences Mehdi Dianatpour Shiraz University of Medical Sciences Kamran Bagheri Lankarani Shiraz University of Medical Sciences Research Keywords: Tamoxifen, breast cancer, genetic variants, RNA-seq. Posted Date: August 17th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-783422/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/33 Abstract Background Breast cancer is one of the most important causes of mortality in the world, and Tamoxifen therapy is known as a medication strategy for estrogen receptor-positive breast cancer. In current study, two hypotheses of Tamoxifen consumption in breast cancer cell line (MCF7) were investigated. First, the effect of Tamoxifen on genes expression prole at transcriptome level was evaluated between the control and treated samples. Second, due to the fact that Tamoxifen is known as a mutagenic factor, there may be an association between the alterations of genetic variants and Tamoxifen treatment, which can impact on the drug response. Methods In current study, the whole-transcriptome (RNA-seq) dataset of four investigations (19 samples) were derived from European Bioinformatics Institute (EBI). At transcriptome level, the effect of Tamoxifen was investigated on gene expression prole between control and treatment samples. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

Mouse Skull Mean Shape and Shape Robustness Rely on Different
Mouse Skull Mean Shape and Shape Robustness Rely on Different Genetic Architectures and Different Loci Ceferino Varón-González, Luisa Pallares, Vincent Debat, Nicolas Navarro To cite this version: Ceferino Varón-González, Luisa Pallares, Vincent Debat, Nicolas Navarro. Mouse Skull Mean Shape and Shape Robustness Rely on Different Genetic Architectures and Different Loci. Frontiers inGe- netics, Frontiers, 2019, 10, pp.64. 10.3389/fgene.2019.00064. hal-02049215 HAL Id: hal-02049215 https://hal.sorbonne-universite.fr/hal-02049215 Submitted on 26 Feb 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. fgene-10-00064 February 9, 2019 Time: 17:37 # 1 ORIGINAL RESEARCH published: 12 February 2019 doi: 10.3389/fgene.2019.00064 Mouse Skull Mean Shape and Shape Robustness Rely on Different Genetic Architectures and Different Loci Ceferino Varón-González1,2, Luisa F. Pallares3, Vincent Debat1† and Nicolas Navarro2,4*† 1 Institut de Systématique, Évolution, Biodiversité, ISYEB – UMR 7205 – CNRS, MNHN, UPMC, EPHE, UA, Muséum National d’Histoire Naturelle, Sorbonne Universités, Paris, France, 2 Biogéosciences, UMR 6282 CNRS, Université Bourgogne Franche-Comté, Dijon, France, 3 Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ, United States, 4 EPHE, PSL University, Dijon, France The genetic architecture of skull shape has been extensively studied in mice and the results suggest a highly polygenic and additive basis. -

Comparative Map Alignment of BTA27 and HSA4 and 8 to Identify Conserved Segments of Genome Containing Fat Deposition QTL Tad S
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by UNL | Libraries University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Roman L. Hruska U.S. Meat Animal Research U.S. Department of Agriculture: Agricultural Center Research Service, Lincoln, Nebraska 2000 Comparative map alignment of BTA27 and HSA4 and 8 to identify conserved segments of genome containing fat deposition QTL Tad S. Sonstegard USDA-ARS Gene Evaluation and Mapping Laboratory, Beltsville Agricultural Research Center Wes M. Garrett USDA-ARS, [email protected] Melissa S. Ashwell USDA-ARS Gene Evaluation and Mapping Laboratory, Beltsville Agricultural Research Center Gary L. Bennett USDA-ARS, [email protected] Steven M. Kappes USDA-ARS See next page for additional authors Follow this and additional works at: http://digitalcommons.unl.edu/hruskareports Sonstegard, Tad S.; Garrett, Wes M.; Ashwell, Melissa S.; Bennett, Gary L.; Kappes, Steven M.; and Van Tassell, Curtis P., "Comparative map alignment of BTA27 and HSA4 and 8 to identify conserved segments of genome containing fat deposition QTL" (2000). Roman L. Hruska U.S. Meat Animal Research Center. 427. http://digitalcommons.unl.edu/hruskareports/427 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Agricultural Research Service, Lincoln, Nebraska at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Roman L. Hruska U.S. Meat Animal Research Center by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Tad S. Sonstegard, Wes M. Garrett, Melissa S. Ashwell, Gary L. -
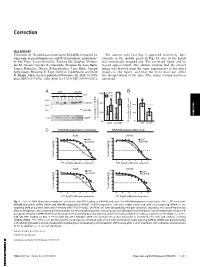
Correction for Zhou Et Al., Scaffold Association Factor B (SAFB) Is Required for Expression of Prenyltransferases and RAS Membra
Correction CELL BIOLOGY Correction for “Scaffold association factor B (SAFB) is required for The authors note that Fig. 5 appeared incorrectly. Spe- expression of prenyltransferases and RAS membrane association,” cifically, in the middle panel of Fig. 5A, one of the bands by Mo Zhou, Leena Kuruvilla, Xiarong Shi, Stephen Viviano, was erroneously cropped out. The corrected figure and its Ian M. Ahearn, Caroline R. Amendola, Wenjuan Su, Sana Badri, legend appear below. The authors confirm that the correct James Mahaffey, Nicole Fehrenbacher, Jane Skok, Joseph image was derived from the same experiments as the other Schlessinger, Benjamin E. Turk, David A. Calderwood, and Mark images in the figure, and that the error does not affect R. Philips, which was first published November 30, 2020; 10.1073/ the interpretation of the data. The online version has been pnas.2005712117 (Proc. Natl. Acad. Sci. U.S.A. 117,31914–31922). corrected. A shNT shSAFB * B 60% - + - + FTI S P S P S P S P Total Ras 40% 100% ** RhoGDI 20% 50% CORRECTION CIMPR in cytosolic fraction FTI treated vs untreated vs untreated FTI treated percentage of total Ras percentage 0% 0% shNT shSAFB GTP-loaded Ras in A549 stables, shNT shSAFB C KRAS4B-dependent lung tumor KRAS4B-independent lung A549 H1437 %cell viability %cell viability FTI (log10, µM) dose response FTI (log10, µM) dose response H358 H1975 %cell viability %cell viability FTI (log10, µM) dose response FTI (log10, µM) dose response Fig. 5. Loss of SAFB diminishes membrane association and GTP loading of KRAS4B and sensitizes KRAS4B-dependent lung tumor cells to FTI treatment. -

Biomedical Informatics
BIOMEDICAL INFORMATICS Abstract GENE LIST AUTOMATICALLY DERIVED FOR YOU (GLAD4U): DERIVING AND PRIORITIZING GENE LISTS FROM PUBMED LITERATURE JEROME JOURQUIN Thesis under the direction of Professor Bing Zhang Answering questions such as ―Which genes are related to breast cancer?‖ usually requires retrieving relevant publications through the PubMed search engine, reading these publications, and manually creating gene lists. This process is both time-consuming and prone to errors. We report GLAD4U (Gene List Automatically Derived For You), a novel, free web-based gene retrieval and prioritization tool. The quality of gene lists created by GLAD4U for three Gene Ontology terms and three disease terms was assessed using ―gold standard‖ lists curated in public databases. We also compared the performance of GLAD4U with that of another gene prioritization software, EBIMed. GLAD4U has a high overall recall. Although precision is generally low, its prioritization methods successfully rank truly relevant genes at the top of generated lists to facilitate efficient browsing. GLAD4U is simple to use, and its interface can be found at: http://bioinfo.vanderbilt.edu/glad4u. Approved ___________________________________________ Date _____________ GENE LIST AUTOMATICALLY DERIVED FOR YOU (GLAD4U): DERIVING AND PRIORITIZING GENE LISTS FROM PUBMED LITERATURE By Jérôme Jourquin Thesis Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Biomedical Informatics May, 2010 Nashville, Tennessee Approved: Professor Bing Zhang Professor Hua Xu Professor Daniel R. Masys ACKNOWLEDGEMENTS I would like to express profound gratitude to my advisor, Dr. Bing Zhang, for his invaluable support, supervision and suggestions throughout this research work. -
Evaluation of Variability in High Resolution Protein Structures by Global Distance Scoring
bioRxiv preprint doi: https://doi.org/10.1101/202028; this version posted December 16, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Evaluation of variability in high resolution protein structures by global distance scoring Risa Anzai, Yoshiki Asami, Waka Inoue, Hina Ueno, Koya Yamada, Tetsuji Okada Department of Life Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan. Corresponding author: Tetsuji Okada, Department of Life Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan. +81-3-3986-0221, [email protected] Abstract Systematic analysis of statistical and dynamical properties of proteins is critical to understanding cellular events. Extraction of biologically relevant information from a set of high-resolution structures is important because it can provide mechanistic details behind the functional properties of protein families, enabling rational comparison between families. Most of the current structure comparisons are pairwise-based, which hampers the global analysis of increasing contents in the Protein Data Bank. Additionally, pairing of protein structures introduces uncertainty with respect to reproducibility because it frequently accompanies other settings for superimposition. This study introduces intramolecular distance scoring, for the analysis of human proteins, for each of which at least several high-resolution are available. We show that the results are comprehensively used to overview advances at the atomic level exploration of each protein and protein family. -

Genomic Landscape of Metastatic Colorectal Cancer
ARTICLE Received 22 Feb 2014 | Accepted 2 Oct 2014 | Published 14 Nov 2014 DOI: 10.1038/ncomms6457 OPEN Genomic landscape of metastatic colorectal cancer Josien C. Haan1, Mariette Labots2, Christian Rausch1, Miriam Koopman3, Jolien Tol4, Leonie J.M. Mekenkamp5, Mark A. van de Wiel6, Danielle Israeli1, Hendrik F. van Essen1, Nicole C.T. van Grieken1, Quirinus J.M. Voorham1, Linda J.W. Bosch1, Xueping Qu7, Omar Kabbarah7, Henk M.W. Verheul2, Iris D. Nagtegaal5, Cornelis J.A. Punt8, Bauke Ylstra1,* & Gerrit A. Meijer1,* Response to drug therapy in individual colorectal cancer (CRC) patients is associated with tumour biology. Here we describe the genomic landscape of tumour samples of a homogeneous well-annotated series of patients with metastatic CRC (mCRC) of two phase III clinical trials, CAIRO and CAIRO2. DNA copy number aberrations of 349 patients are determined. Within three treatment arms, 194 chromosomal subregions are associated with progression-free survival (PFS; uncorrected single-test P-values o0.005). These subregions are filtered for effect on messenger RNA expression, using an independent data set from The Cancer Genome Atlas which returned 171 genes. Three chromosomal regions are associated with a significant difference in PFS between treatment arms with or without irinotecan. One of these regions, 6q16.1–q21, correlates in vitro with sensitivity to SN-38, the active metabolite of irinotecan. This genomic landscape of mCRC reveals a number of DNA copy number aberrations associated with response to drug therapy. 1 Department of Pathology, VU University Medical Center, PO Box 7057, 1007 MB Amsterdam, The Netherlands. 2 Department of Medical Oncology, VU University Medical Center, PO Box 7057, 1007 MB Amsterdam, The Netherlands. -

A Gene for Universal Congenital Alopecia Maps to Chromosome 8P21-22 Markus M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Am. J. Hum. Genet. 62:386–390, 1998 A Gene for Universal Congenital Alopecia Maps to Chromosome 8p21-22 Markus M. No¨then,1 Sven Cichon,1 Ina R. Vogt,1 Susanne Hemmer,1 Roland Kruse,1 Michael Knapp,2 Tobias Ho¨ller,2 Muhammad Faiyaz ul Haque,4,5 Sayedul Haque,4 Peter Propping,1 Mahmud Ahmad,4 and Marcella Rietschel1,3 1Institute of Human Genetics, 2Institute for Medical Statistics, and 3Department of Psychiatry, University of Bonn, Bonn; 4Department of Biological Sciences, Quaid-i-Azam University, Islamabad, Pakistan; and 5Medical Genetics, Ahmanson Department of Pediatrics, Cedars-Sinai Research Institute, Los Angeles Summary alopecia may occur either in isolation or with associated defects. On the basis of such associations, several dif- Complete or partial congenital absence of hair (congen- ferent syndromes featuring congenital alopecia can be ital alopecia) may occur either in isolation or with as- distinguished (Birnbaum and Baden 1987; Feinstein et sociated defects. The majority of families with isolated al. 1987; Vogt et al. 1988). The isolated form of con- congenital alopecia has been reported to follow an au- genital alopecia has been reported in sporadic and fa- tosomal-recessive mode of inheritance (MIM 203655). milial cases. In familial cases, inheritance is usu- As yet, no gene has been linked to isolated congenital ally autosomal recessive (MIM 203655 [http:// alopecia, nor has linkage been established to a specific www3.ncbi.nlm.nih.gov:80/htbin-post/Omim/dispmim? region of the genome. -

Analysis of Protein-Protein Interaction Byin Vivo Quantitative Proteomics In
Analysis of protein-protein interaction by in vivo quantitative proteomics in Caenorhabditis elegans D i s s e r t a t i o n zur Erlangung des akademischen Grades d o c t o r r e r u m n a t u r a l i u m (Dr. rer. nat.) im Fach Biologie eingereicht an der Lebenswissenschaftlichen Fakultät der Humboldt-Universität zu Berlin von M.Sc. Jiaxuan Chen Präsident der Humboldt-Universität zu Berlin Prof. Dr. Jan-Hendrik Olbertz Dekan der Lebenswissenschaftlichen Fakultät Prof. Dr. Richard Lucius Gutachter/innen: 1. Prof. Dr. Richard Lucius 2. Prof. Dr. Matthias Selbach 3. Dr. Markus Landthaler Tag der mündlichen Prüfung: 21.09.2015 for my parents “No man is an island, entire of itself.” John Donne Table of Contents Table of Contents ................................................................................................................. I Abstract ............................................................................................................................... V Zusammenfassung ............................................................................................................ VI I Introduction ................................................................................................................ 7 I.1. Protein-protein interaction ........................................................................................................ 7 I.1.1. Detection of protein-protein interactions ................................................................................. 8 I.1.2. Affinity purification and mass spectrometry