Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MLR) Model and Filtration
SUPPLEMENT Contents 1. Extended description of the Data sets. 2. Extended description of the multiple linear regression (MLR) model and filtration. 3. Extended description of the random-gene classifiers. 4. Extended description of the comparison with Dakhova et al. (1)and Richardson et al. (2) 5. Extended description of preparation of RNA and the XP_PCR protocol. 6. Table S1. Comparison of 131-probe set Diagnostic Classifier to classifiers generated with ‘random’ genes. 7. Table S2. Concordance of 38 overlapping genes/probe sets of the 339 probe sets ( basis) of the Diagnostic Classifier with the sign of differential change of Dakhova et al. (1). 8. Table S3. Function enrichment analysis. 9. Table S4. PCR validation of preferential expression in stroma by representative genes of the Diagnostic Classifier. 10. Figure S1. The incidence numbers of 339 probe sets obtained by 105-fold permutation procedure for gene selection. 11. Figure S2. Heatmap using the Diagnostic Classifier to categorize all training cases. 12. Figure S3. Heatmap of all 364 test samples used in this study as categorized by the 131 probe set Diagnostic Classifier. 13. Figure S4. Cluster diagram of the cases of Dakhova et al. (1) using only the 38 overlapping genes. 14. References for the Supplement. 1 1. Extended description of the Data Sets. Datasets 1 and 2 (Table 1) are based on post-prostatectomy frozen tissue samples obtained by informed consent using IRB-approved and HIPPA-compliant protocols. All tissues, except where noted, were collected at surgery and escorted to pathology for expedited review, dissection, and snap freezing in liquid nitrogen. -

Supplemental Data
MOLECULAR PHARMACOLOGY Supplemental Data Regulation of M3 Muscarinic Receptor Expression and Function by Transmembrane Protein 147 Erica Rosemond, Mario Rossi, Sara M. McMillin, Marco Scarselli, Julie G. Donaldson, and Jürgen Wess Supplemental Table 1 M3R-interacting proteins identified in a membrane-based yeast two-hybrid screen Accession Protein No. Tetraspanin family CD82 AAB23825 CD9 P21926 CD63 AAV38940 Ubiquitin-associated proteins Small ubiquitin-related modifier 2 precursor (SUMO-1) AAC50996 Small ubiquitin-related modifier 2 precursor (SUMO-2) P61956 UBC protein AAH08955 Ubiquitin C NP_066289 Receptor proteins/Transmembrane proteins AAH01118 / Transmembrane protein 147 BC001118 G protein-coupled receptor 37 NP_005293 NM_016235 / Homo sapiens G protein-coupled receptor, family C, group 5, member B (GPRC5B) AAF05331 Rhodopsin NP_000530 Aquaporin-4 (AQP-4) P55087 Glutamate receptor, ionotropic, N-methyl D-aspartate-associated protein 1 NP_001009184 Sodium channel, voltage gated, type VIII, alpha NP_055006 Transmembrane 9 superfamily member 2 CAH71381 Transmembrane protein 14A AAH19328 Signaling molecules Phosphatidic acid phosphatase type 2C AAP35667 Calmodulin 2 AAH08437 Protein kinase Njmu-R1 AAH54035 2',3'-Cyclic nucleotide 3' phosphodiesterase (CNP) AAH06392 Solute carrier proteins Solute carrier family 39 (zinc transporter), member 3 isoform a NP_653165 Solute carrier family 22 (organic cation transporter), member 17 isoform b NP_057693 Solute carrier family 31 (copper transporters), member 2 NP_001851 Solute carrier family -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Translational Research of the Quaking Gene
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1381 Translational research of the quaking gene Focusing on the conjunction between development and disease BRYN FARNSWORTH ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-554-9595-4 UPPSALA urn:nbn:se:uu:diva-287408 2016 Dissertation presented at Uppsala University to be publicly examined in Zootisalen, EBC, Norbyvägen 18A, Uppsala, Tuesday, 14 June 2016 at 13:00 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Dr. Roman Chrast (Department of Neuroscience, Karolinska University). Abstract Farnsworth, B. 2016. Translational research of the quaking gene. Focusing on the conjunction between development and disease. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1381. 61 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9595-4. Quaking (QKI) is an RNA binding protein involved in the post-transcriptional regulation of gene expression. Originally identified as the cause of hypomyelination in a mouse mutant, it has since been consistently implicated in a wide range of neurological diseases. As a gene exclusively expressed in glial cells of the central nervous system, such associations emphasise the importance of an indirect, or non-neuronal link to aberrant neural function. A role in early neural development has also been suggested from the viable and embryonic lethal mouse mutants, yet detailed and in vivo study has been precluded thus far by the murine uterine gestation, and mutant lethality prior to oligodendrogenesis. This thesis examines the role of QKI in human neurological disease, and explores the use of the zebrafish as a model organism to allow the unimpeded study of neural development. -

Pathological Relationships Involving Iron and Myelin May Constitute a Shared Mechanism Linking Various Rare and Common Brain Diseases
Rare Diseases ISSN: (Print) 2167-5511 (Online) Journal homepage: http://www.tandfonline.com/loi/krad20 Pathological relationships involving iron and myelin may constitute a shared mechanism linking various rare and common brain diseases Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna F. Collingwood, Conceição Bettencourt, Henry Houlden, Mina Ryten , John K. Olynyk, Debbie Trinder, Daniel M. Johnstone & Elizabeth A. Milward To cite this article: Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna F. Collingwood, Conceição Bettencourt, Henry Houlden, Mina Ryten , John K. Olynyk, Debbie Trinder, Daniel M. Johnstone & Elizabeth A. Milward (2016) Pathological relationships involving iron and myelin may constitute a shared mechanism linking various rare and common brain diseases, Rare Diseases, 4:1, e1198458, DOI: 10.1080/21675511.2016.1198458 To link to this article: http://dx.doi.org/10.1080/21675511.2016.1198458 © 2016 The Author(s). Published with View supplementary material license by Taylor & Francis Group, LLC© Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna Accepted author version posted online: 22 Submit your article to this journal JunF. Collingwood, 2016. Conceição Bettencourt, PublishedHenry Houlden, online: Mina 22 Jun Ryten, 2016. for the UK Brain Expression Consortium (UKBEC), John K. Olynyk, Debbie Trinder, Daniel M. Johnstone,Article views: and 541 Elizabeth A. Milward. View related articles View Crossmark data Citing articles: 2 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=krad20 Download by: [University of Newcastle, Australia] Date: 17 May 2017, At: 19:57 RARE DISEASES 2016, VOL. -

PLP1 Gene Proteolipid Protein 1
PLP1 gene proteolipid protein 1 Normal Function The PLP1 gene provides instructions for producing proteolipid protein 1 and a modified version (isoform) of that protein called DM20. Proteolipid protein 1 is found primarily in nerves in the brain and spinal cord (the central nervous system) and DM20 is produced mainly in nerves that connect the brain and spinal cord to muscles (the peripheral nervous system). These two proteins are found within the cell membrane of nerve cells, where they make up a large proportion of myelin and help myelin stay anchored to the cells. Myelin is the fatty covering that insulates nerve fibers and promotes the rapid transmission of nerve impulses. Health Conditions Related to Genetic Changes Pelizaeus-Merzbacher disease There are more than 280 mutations in the PLP1 gene that have been found to cause Pelizaeus-Merzbacher disease. Pelizaeus-Merzbacher disease is an inherited condition involving the central nervous system that primarily affects males. Individuals with Pelizaeus-Merzbacher disease have neurological problems including abnormal eye movements (nystagmus) and other movement abnormalities. In addition, these individuals have difficulty walking or cannot walk. An extra copy (duplication) of the PLP1 gene accounts for 50 to 70 percent of all Pelizaeus-Merzbacher disease mutations. In many cases, genes near the PLP1 gene are also duplicated, but having extra copies of these genes does not seem to impact the severity of the condition. In another 10 to 25 percent of cases, mutations change single protein building blocks (amino acids) in the proteolipid protein 1 and DM20 proteins and lead to excess or abnormal proteins that are often misfolded. -

Quantitative Multiplex Real-Time PCR for Detection of PLP1 Gene Duplications in Pelizaeus–Merzbacher Patients
GENETIC TESTING Volume 10, Number 3, 2006 © Mary Ann Liebert, Inc. Quantitative Multiplex Real-Time PCR for Detection of PLP1 Gene Duplications in Pelizaeus–Merzbacher Patients EMILIE MIKESˇOVÁ,1 LUCIA BARÁNKOVÁ,2 IVA SAKMARYOVÁ,1 IVA TATARKOVÁ,1 and PAVEL SEEMAN1 ABSTRACT Pelizaeus–Merzbacher disease (PMD) is an X-linked recessive disorder of central nervous system (CNS) myeli- nation typically affecting males. A genomic duplication of variable size at Xq22.2, containing the entire pro- teolipid protein 1 gene (PLP1), is responsible for approximately 60–70% of PMD cases. The aim of this study was to develop a rapid and robust method for determination of PLP1 gene dosage. We optimized two multi- plex real-time quantitative PCR (Q-PCR) assays targeting exons 3 and 6 of the PLP1 gene, and then validated these assays by retrospective analysis of a set of genomic DNAs from 67 previously tested patients and 43 nor- mal controls. Samples were analyzed in multiplex PCR reactions using TaqMan chemistry and the ABI Prism 7000 Sequence Detection System. PLP1 dosage was determined by the relative quantitative comparative thresh- old cycle method (⌬⌬Ct) using the human serum albumin gene as the endogenous reference gene. Three clearly non-overlapping ranges of results, corresponding to the presence of one, two, or three PLP1 copies, were de- tected in both assays. The results were completely concordant with gender and previous PLP1 gene dosage testing based on quantitative fluorescent multiplex PCR and analysis of a dinucleotide polymorphism in the first intron of the PLP1 gene. We conclude that multiplex real-time Q-PCR represents a fast and reliable tool for PLP1 duplication testing in PMD families. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Supplemental Table S1. Primers for Sybrgreen Quantitative RT-PCR Assays
Supplemental Table S1. Primers for SYBRGreen quantitative RT-PCR assays. Gene Accession Primer Sequence Length Start Stop Tm GC% GAPDH NM_002046.3 GAPDH F TCCTGTTCGACAGTCAGCCGCA 22 39 60 60.43 59.09 GAPDH R GCGCCCAATACGACCAAATCCGT 23 150 128 60.12 56.52 Exon junction 131/132 (reverse primer) on template NM_002046.3 DNAH6 NM_001370.1 DNAH6 F GGGCCTGGTGCTGCTTTGATGA 22 4690 4711 59.66 59.09% DNAH6 R TAGAGAGCTTTGCCGCTTTGGCG 23 4797 4775 60.06 56.52% Exon junction 4790/4791 (reverse primer) on template NM_001370.1 DNAH7 NM_018897.2 DNAH7 F TGCTGCATGAGCGGGCGATTA 21 9973 9993 59.25 57.14% DNAH7 R AGGAAGCCATGTACAAAGGTTGGCA 25 10073 10049 58.85 48.00% Exon junction 9989/9990 (forward primer) on template NM_018897.2 DNAI1 NM_012144.2 DNAI1 F AACAGATGTGCCTGCAGCTGGG 22 673 694 59.67 59.09 DNAI1 R TCTCGATCCCGGACAGGGTTGT 22 822 801 59.07 59.09 Exon junction 814/815 (reverse primer) on template NM_012144.2 RPGRIP1L NM_015272.2 RPGRIP1L F TCCCAAGGTTTCACAAGAAGGCAGT 25 3118 3142 58.5 48.00% RPGRIP1L R TGCCAAGCTTTGTTCTGCAAGCTGA 25 3238 3214 60.06 48.00% Exon junction 3124/3125 (forward primer) on template NM_015272.2 Supplemental Table S2. Transcripts that differentiate IPF/UIP from controls at 5%FDR Fold- p-value Change Transcript Gene p-value p-value p-value (IPF/UIP (IPF/UIP Cluster ID RefSeq Symbol gene_assignment (Age) (Gender) (Smoking) vs. C) vs. C) NM_001178008 // CBS // cystathionine-beta- 8070632 NM_001178008 CBS synthase // 21q22.3 // 875 /// NM_0000 0.456642 0.314761 0.418564 4.83E-36 -2.23 NM_003013 // SFRP2 // secreted frizzled- 8103254 NM_003013 -

Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication
11788 • The Journal of Neuroscience, July 17, 2013 • 33(29):11788–11799 Neurobiology of Disease Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication Kristi Clark,1,2 Lauren Sakowski,1,2 Karen Sperle,1 Linda Banser,1 Carlisle P. Landel,3 Denise A. Bessert,4 Robert P. Skoff,4 and Grace M. Hobson1,2,5 1Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, Delaware 19803, 2University of Delaware, Department of Biology, Newark, Delaware 19716, 3Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Wayne State University, Department of Anatomy and Cell Biology, Detroit, Michigan 48201, and 5Jefferson Medical College, Thomas Jefferson University, Philadelphia, Pennsylvania 19107 Pelizaeus-Merzbacher disease (PMD) is a hypomyelinating leukodystrophy caused by mutations of the proteolipid protein 1 gene (PLP1), which is located on the X chromosome and encodes the most abundant protein of myelin in the central nervous sytem. Approximately 60% of PMD cases result from genomic duplications of a region of the X chromosome that includes the entire PLP1 gene. The duplications are typically in a head-to-tail arrangement, and they vary in size and gene content. Although rodent models with extra copies of Plp1 have been developed, none contains an actual genomic rearrangement that resembles those found in PMD patients. We used mutagenic insertion chromosome engineering resources to generate the Plp1dup mouse model by introducing an X chromosome duplication in the mouse genome that contains Plp1 and five neighboring genes that are also commonly duplicated in PMD patients. The Plp1dup mice display progressive gait abnormalities compared with wild-type littermates. -

Tandem Duplications of Two Separate Fragments of the Dystrophin Gene in a Patient with Duchenne Muscular Dystrophy
J Hum Genet (2008) 53:215–219 DOI 10.1007/s10038-007-0235-1 ORIGINAL ARTICLE Tandem duplications of two separate fragments of the dystrophin gene in a patient with Duchenne muscular dystrophy Zhujun Zhang Æ Yasuhiro Takeshima Æ Hiroyuki Awano Æ Atsushi Nishiyama Æ Yo Okizuka Æ Mariko Yagi Æ Masafumi Matsuo Received: 7 November 2007 / Accepted: 3 December 2007 / Published online: 27 December 2007 Ó The Japan Society of Human Genetics and Springer 2007 Abstract Mutations in the dystrophin gene result in the Introduction most common inherited muscle disease, Duchenne mus- cular dystrophy (DMD). Duplications spanning one or Duchenne muscular dystrophy (DMD) is the most common more exons have been found to be the second most com- inherited muscle disease, affecting 1 in every 3,500 male mon disease-causing mutation in the dystrophin gene. births. This disease is caused by mutations in the dystro- Although the duplicated exons are commonly thought to be phin gene located on Xp21. Deletions involving one or arranged in tandem, rare noncontiguous exon duplications more exons are the most common type of mutation asso- have been disclosed without clarifying their location or ciated with DMD, accounting for nearly two-thirds of all orientation. Here we present the first report that details the cases. Duplications are the second most common type of exact locations and orientations of noncontiguous dupli- mutation in this gene, occurring in approximately 5–10% cations in the dystrophin gene. Multiplex ligation- of DMD patients (Hu et al. 1990; White et al. 2002, 2006). dependent probe amplification analysis of the dystrophin Because the dystrophin gene consists of 79 exons gene of a Japanese boy with DMD revealed that his spanning more than 2,500 kb in the human genome, it has genomic DNA contained duplications of exons from two been difficult to examine every exon for deletions or separate fragments of the gene: one from exon 45 to exon duplications. -
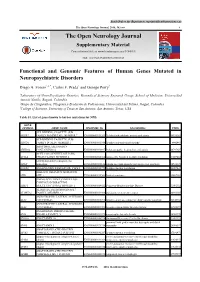
The Open Neurology Journal, 2016, 10, I-Xv I the Open Neurology Journal Supplementary Material Content List Available At
Send Orders for Reprints to [email protected] The Open Neurology Journal, 2016, 10, i-xv i The Open Neurology Journal Supplementary Material Content list available at: www.benthamopen.com/TONEUJ/ DOI: 10.2174/1874205X01610010143 Functional and Genomic Features of Human Genes Mutated in Neuropsychiatric Disorders Diego A. Forero1,4, *, Carlos F. Prada2 and George Perry3 1Laboratory of NeuroPsychiatric Genetics, Biomedical Sciences Research Group, School of Medicine, Universidad Antonio Nariño. Bogotá, Colombia 2Grupo de Citogenética, Filogenia y Evolución de Poblaciones, Universidad del Tolima. Ibagué, Colombia 3College of Sciences, University of Texas at San Antonio, San Antonio, Texas, USA Table S1. List of genes known to harbor mutations for NPD. GENE SYMBOL GENE NAME ENSEMBL ID DISORDERS PMID ATP-BINDING CASSETTE, SUB- ABCB7 FAMILY B (MDR/TAP), MEMBER 7 ENSG00000131269 X-linked sideroblastic anemia and ataxia 10196363 ATP-BINDING CASSETTE, SUB- ABCD1 FAMILY D (ALD), MEMBER 1 ENSG00000101986 X-linked adrenoleukodystrophy 8441467 ABHYDROLASE DOMAIN ABHD12 CONTAINING 12 ENSG00000100997 Polyneuropathy, hearing loss and ataxia 20797687 ACYL-COA SYNTHETASE LONG- ACSL4 CHAIN FAMILY MEMBER 4 ENSG00000068366 nonspecific X-linked mental retardation 11889465 activity-dependent neuroprotector ADNP homeobox ENSG00000101126 autism spectrum disorder and intellectual disability 24531329 AGTR2 ANGIOTENSIN II RECEPTOR, TYPE 2 ENSG00000180772 X-linked mental retardation 12089445 ABELSON HELPER INTEGRATION AHI1 SITE 1 ENSG00000135541 Joubert