University of Cincinnati
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -

Detailed Review Paper on Retinoid Pathway Signalling
1 1 Detailed Review Paper on Retinoid Pathway Signalling 2 December 2020 3 2 4 Foreword 5 1. Project 4.97 to develop a Detailed Review Paper (DRP) on the Retinoid System 6 was added to the Test Guidelines Programme work plan in 2015. The project was 7 originally proposed by Sweden and the European Commission later joined the project as 8 a co-lead. In 2019, the OECD Secretariat was added to coordinate input from expert 9 consultants. The initial objectives of the project were to: 10 draft a review of the biology of retinoid signalling pathway, 11 describe retinoid-mediated effects on various organ systems, 12 identify relevant retinoid in vitro and ex vivo assays that measure mechanistic 13 effects of chemicals for development, and 14 Identify in vivo endpoints that could be added to existing test guidelines to 15 identify chemical effects on retinoid pathway signalling. 16 2. This DRP is intended to expand the recommendations for the retinoid pathway 17 included in the OECD Detailed Review Paper on the State of the Science on Novel In 18 vitro and In vivo Screening and Testing Methods and Endpoints for Evaluating 19 Endocrine Disruptors (DRP No 178). The retinoid signalling pathway was one of seven 20 endocrine pathways considered to be susceptible to environmental endocrine disruption 21 and for which relevant endpoints could be measured in new or existing OECD Test 22 Guidelines for evaluating endocrine disruption. Due to the complexity of retinoid 23 signalling across multiple organ systems, this effort was foreseen as a multi-step process. -
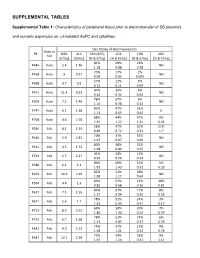
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Topoisomerase Ii Inhibitors Induce an Illegitimate Genome Rearrangement Common in Infant Leukemia
TOPOISOMERASE II INHIBITORS INDUCE AN ILLEGITIMATE GENOME REARRANGEMENT COMMON IN INFANT LEUKEMIA by Bhawana Bariar A dissertation submitted to the faculty of The University of North Carolina at Charlotte in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology Charlotte 2013 Approved by: ______________________________ Dr. Christine Richardson ______________________________ Dr. Mark Clemens ______________________________ Dr. Laura Schrum ______________________________ Dr. Pinku Mukherjee ______________________________ Dr. Anthony Fodor ii ©2013 Bhawana Bariar ALL RIGHTS RESERVED iii ABSTRACT BHAWANA BARIAR. Topoisomerase II inhibitors induce an illegitimate genome rearrangement common in infant leukemia. (Under the direction of DR. CHRISTINE RICHARDSON) Infant acute leukemias account for ~30% of all malignancy seen in childhood across the Western world. They are aggressive and characterized by rapid onset shortly after birth. The majority of these have rearrangements involving the MLL (mixed lineage leukemia) gene. Although MLL fusion to more than 75 genes have been identified, AF9 is one of its most common translocation partners. Since MLL breakpoint sequences associated with infant acute leukemia are similar to those in secondary AML following exposure to the topoisomerase II (topo II) poison etoposide, it has been hypothesized that exposure during pregnancy to biochemically similar compounds may promote infant acute leukemia. Some studies have shown an epidemiological link between bioflavonoid intake -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Piggybac Mutagenesis and Exome Sequencing Identify Genetic Driver
Noorani et al. Genome Biology (2020) 21:181 https://doi.org/10.1186/s13059-020-02092-2 RESEARCH Open Access PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas Imran Noorani1,2* , Jorge de la Rosa1†, Yoon Ha Choi1,3†, Alexander Strong1, Hannes Ponstingl1, M. S. Vijayabaskar1, Jusung Lee3, Eunmin Lee3, Angela Richard-Londt4, Mathias Friedrich1,5, Federica Furlanetto5, Rocio Fuente1, Ruby Banerjee1, Fengtang Yang1, Frances Law1, Colin Watts2,6, Roland Rad5, George Vassiliou1, Jong Kyoung Kim3, Thomas Santarius2, Sebastian Brandner4 and Allan Bradley1* * Correspondence: [email protected]; [email protected] Abstract †Jorge de la Rosa and Yoonha Choi contributed equally to this work. Background: Glioma is the most common intrinsic brain tumor and also occurs in 1The Wellcome Trust Sanger the spinal cord. Activating EGFR mutations are common in IDH1 wild-type gliomas. Institute, Wellcome Trust Genome However, the cooperative partners of EGFR driving gliomagenesis remain poorly Campus, Hinxton, Cambridgeshire CB10 1SA, UK understood. Full list of author information is Results: We explore EGFR-mutant glioma evolution in conditional mutant mice by available at the end of the article whole-exome sequencing, transposon mutagenesis forward genetic screening, and transcriptomics. We show mutant EGFR is sufficient to initiate gliomagenesis in vivo, both in the brain and spinal cord. We identify significantly recurrent somatic alterations in these gliomas including mutant EGFR amplifications and Sub1, Trp53, and Tead2 loss-of-function mutations. Comprehensive functional characterization of 96 gliomas by genome-wide piggyBac insertional mutagenesis in vivo identifies 281 known and novel EGFR-cooperating driver genes, including Cdkn2a, Nf1, Spred1, and Nav3. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone. -

Creb5 Establishes the Competence for Prg4 Expression in Articular Cartilage
ARTICLE https://doi.org/10.1038/s42003-021-01857-0 OPEN Creb5 establishes the competence for Prg4 expression in articular cartilage Cheng-Hai Zhang1, Yao Gao1, Unmesh Jadhav2,3, Han-Hwa Hung4, Kristina M. Holton5, Alan J. Grodzinsky4, ✉ Ramesh A. Shivdasani 2,3,6 & Andrew B. Lassar 1 A hallmark of cells comprising the superficial zone of articular cartilage is their expression of lubricin, encoded by the Prg4 gene, that lubricates the joint and protects against the devel- opment of arthritis. Here, we identify Creb5 as a transcription factor that is specifically expressed in superficial zone articular chondrocytes and is required for TGF-β and EGFR signaling to induce Prg4 expression. Notably, forced expression of Creb5 in chondrocytes 1234567890():,; derived from the deep zone of the articular cartilage confers the competence for TGF-β and EGFR signals to induce Prg4 expression. Chromatin-IP and ATAC-Seq analyses have revealed that Creb5 directly binds to two Prg4 promoter-proximal regulatory elements, that display an open chromatin conformation specifically in superficial zone articular chondrocytes; and which work in combination with a more distal regulatory element to drive induction of Prg4 by TGF-β. Our results indicate that Creb5 is a critical regulator of Prg4/lubricin expression in the articular cartilage. 1 Department of Biological Chemistry and Molecular Pharmacology, Blavatnik Institute at Harvard Medical School, Boston, MA, USA. 2 Department of Medical Oncology and Center for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, MA, USA. 3 Departments of Medicine, Brigham & Women’s Hospital and Harvard Medical School, Boston, MA, USA. 4 Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Essential Role of Autophagy in Protecting Neonatal Haematopoietic Stem Cells from Oxidative Stress in a P62-Independent Manner
www.nature.com/scientificreports OPEN Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62‑independent manner Naho Nomura1,7,10, Chiaki Ito1,10, Takako Ooshio1,8, Yuko Tadokoro1,2, Susumu Kohno3, Masaya Ueno1,2, Masahiko Kobayashi1,2, Atsuko Kasahara4, Yusuke Takase1,9, Kenta Kurayoshi1, Sha Si1,2, Chiaki Takahashi3, Masaaki Komatsu5, Toru Yanagawa6 & Atsushi Hirao1,2* Autophagy is a cellular degradation system contributing to homeostasis of tissue stem cells including haematopoietic stem cells (HSCs). It plays pleiotropic roles in HSC characteristics throughout life, but its stage‑specifc roles in HSC self‑renewal are unclear. To investigate the efects of Atg5 deletion on stage‑specifc HSC functions, we compared the repopulating capacity of HSCs in Atg5f/f;Vavi-cre mice from postnatal day (P) 0–7 weeks of age. Interestingly, Atg5 defciency led to no remarkable abnormality in the HSC self‑renewal capacity at P0, but signifcant defects at P7, followed by severe defects. Induction of Atg5 deletion at P5 by tamoxifen administration to Atg5f/f;Rosa26-Cre-ERT2 mice resulted in normal haematopoiesis, including the HSC population, until around 1 year, suggesting that Atg5 in the early neonatal period was critical for haematopoiesis in adults. Mitochondrial oxidative stress was increased by Atg5 loss in neonatal HSC/progenitor cells. Although p62 had accumulated in immature bone marrow cells of Atg5f/f;Vavi-cre mice, p62 deletion did not restore defective HSC functions, indicating that Atg5‑dependent haematopoietic regulation in the developmental period was independent of p62. This study proposes a critical role of autophagy in HSC protection against harsh environments in the early neonatal stage, which is essential for healthy long‑term haematopoiesis. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest.