Article Reference
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EYAL AKIVA, Phd
Eyal Akiva CV, Nov. 2017 EYAL AKIVA, PhD Department of Bioengineering and Therapeutic Sciences Phone +1-650-504-9008 University of California at San Francisco Email [email protected] 1700 4th street, San Francisco, Web www.babbittlab.ucsf.edu/eakiva CA, USA EDUCATION 2012-2017 Post-doctoral fellowship at UCSF, Dept. Of Bioengineering and Therapeutic Sciences. Host: Prof. Patricia Babbitt. 2010-2012 Post-doctoral fellowship at UCSF, Dept. Of Bioengineering and Therapeutic Sciences. Host: Prof. Tanja Kortemme. 2004-2010 PhD at The Hebrew University of Jerusalem (Israel), bioinformatics. Host: Prof. Hanah Margalit. “Various Aspects of Modularity in Protein-Protein Interaction". 2001-2004 MSc at The Hebrew University of Jerusalem (Israel), bioinformatics and human genetics. Host: Prof. Muli Ben-Sasson. “Exploiting the Exploiters: Identification of Virus-Host Pep- tide Mimicry as a Source for Modules of Functional Significance”. MAGNA CUM LAUDE. 1997-2000 BSc at Bar-Ilan University (Israel), biology (major) and computer science (minor). Final project advisor: Prof. Ramit Mehr “Modeling the Evolution of the Immune System: a Sim- ulation of the Evolution of Genes that Encode the Variable Regions of Immunoglobulins”. MAGNA CUM LAUDE. 1996-1997 First year of "Industrial Engineering and Management" studies, Tel-Aviv University, Israel. OTHER WORK EXPERIENCE 2000-01 ‘Do-coop technologies’: Team leader and chemistry/microbiology researcher; development of biological applications and manufacture of proprietary nanoparticles (Or Yehuda, Israel and Tel-Aviv University (Prof. Eshel Ben-Jacob’s lab at the school of physics)). FUNDING, HONORS AND AWARDS 2017 Grant: Co-PI, “Utilizing metagenomic sequences for enzyme function prediction”, Joint Genome Institute (US Department of Energy) (http://jgi.doe.gov/doe-user-facilities-ficus- join-forces-to-tackle-biology-big-data/). -

Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by Pickeringhutchins Ltd
European Bioinformatics Institute EMBL-EBI Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by PickeringHutchins Ltd www.pickeringhutchins.com EMBL member states: Austria, Croatia, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, United Kingdom. Associate member state: Australia EMBL-EBI is a part of the European Molecular Biology Laboratory (EMBL) EMBL-EBI EMBL-EBI EMBL-EBI EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus, Hinxton Cambridge CB10 1SD United Kingdom Tel. +44 (0)1223 494 444, Fax +44 (0)1223 494 468 www.ebi.ac.uk EMBL Heidelberg Meyerhofstraße 1 69117 Heidelberg Germany Tel. +49 (0)6221 3870, Fax +49 (0)6221 387 8306 www.embl.org [email protected] EMBL Grenoble 6, rue Jules Horowitz, BP181 38042 Grenoble, Cedex 9 France Tel. +33 (0)476 20 7269, Fax +33 (0)476 20 2199 EMBL Hamburg c/o DESY Notkestraße 85 22603 Hamburg Germany Tel. +49 (0)4089 902 110, Fax +49 (0)4089 902 149 EMBL Monterotondo Adriano Buzzati-Traverso Campus Via Ramarini, 32 00015 Monterotondo (Rome) Italy Tel. +39 (0)6900 91402, Fax +39 (0)6900 91406 © 2012 EMBL-European Bioinformatics Institute All texts written by EBI-EMBL Group and Team Leaders. This publication was produced by the EBI’s Outreach and Training Programme. Contents Introduction Foreword 2 Major Achievements 2011 4 Services Rolf Apweiler and Ewan Birney: Protein and nucleotide data 10 Guy Cochrane: The European Nucleotide Archive 14 Paul Flicek: -

Automated Function Prediction SIG Meeting July 20Th 2013, Berlin, Germany ISMB/ECCB 2013
Automated Function Prediction SIG Meeting July 20th 2013, Berlin, Germany ISMB/ECCB 2013 Keynote Speakers Alex Bateman European Bioinformatics Institute, UK. Using protein domains and families for functional prediction Patricia Babbitt University of California, San Francisco, USA. Protein similarity networks: Identification of functional trends from the context of sequence similarity Anna Tramontano University of Rome, “La Sapienza”, Italy. CASP: aging or growing up? Keith Dunker Indiana University, USA. Functions of Intrinsically Disordered Proteins Automated Function Prediction SIG 2013 Programme 08:30 Welcome from the organisers 08:45 Keynote – Keith Dunker, Indiana University, USA. Functions of Intrinsically Disordered Proteins 09:15 Giorgio Valentini, Università degli Studi di Milano, Italy. Scalable Network-based Learning Methods for Automated Function Prediction based on the Neo4j Graph-database 09:35 Joachim Bargsten, Wageningen University, The Netherlands. Integrated network- and sequence-based protein function prediction across the plant kingdom 09:55 Christos Ouzounis, Institute of Applied Biosciences, Greece. We still haz a job: genome annotashuns 10:15 Coffee break 10:45 Keynote – Patricia Babbitt, University of California, San Francisco, USA. Protein similarity networks: Identification of functional trends from the context of sequence similarity 11:15 Rachael Huntley, European Bioinformatics Institute, UK. The Gene Ontology Resource – Common Misconceptions 11:35 Nives Skunca, ETH Zurich, Switzerland. Assessing protein function predictions in light of the Open World Assumption 11:55 Noah Youngs, New York University, USA. Negative Example Selection in Protein Function Prediction 12:15 Lunch 13:30 Keynote – Anna Tramontano, University of Rome “La Sapienza”, Italy. CASP: aging or growing up? 14:00 Indika Kahanda, Colorado State University, USA. -

AFP / Biosapiens 2008 SIG Meeting Program Toronto, Canada, July 2008
AFP / Biosapiens 2008 SIG Meeting Program Toronto, Canada, July 2008 Dear AFP / Biosapiens 2008 attendee, Welcome to the joint Automated Function Prediction / Biosapiens meeting in ISMB 2008. This is the second time AFP and Biosapiens are joining forces to bring you an engaging and stimulating program. As usual, we strive to bring you the latest in cutting-edge research in computational gene and protein function prediction and annotation delivered by leading international researchers. This year we are holding a joint session with the 3D SIG on predicting function from protein structure. “Structure to Function” is a problem that is rapidly moving to the forefront of life science due to the increasing number of unannotated structures coming from structural genomics projects. We are also addressing a need voiced by the community to learn more about computational function prediction. Kimmen Sjölander from the University of California Berkeley will conduct a workshop on function prediction using phylogenomics. Yanay Ofran from Bar Ilan University, Israel and Predrag Radivojac from Indiana University will present a variety of components, tools and techniques for computational function prediction. Those tutorials are intended for researchers and students that are entering the field, and for those in the field to gain more knowledge and expertise. This is a rare opportunity to interact informally with leading experts in the field and enhance your own research. We would like to thank the members of the Program Committee for carefully reviewing the abstracts sent to this meeting. Thanks to the International Society for Computational Biology for hosting this meeting at ISMB 2008. A special thanks to Prof. -

Reports from the Fifth Edition of CAGI: the Critical Assessment of Genome Interpretation
Received: 16 July 2019 | Accepted: 19 July 2019 DOI: 10.1002/humu.23876 OVERVIEW Reports from the fifth edition of CAGI: The Critical Assessment of Genome Interpretation Gaia Andreoletti1 | Lipika R. Pal2 | John Moult2,3 | Steven E. Brenner1,4 1Department of Plant and Microbial Biology, University of California, Berkeley, California Abstract 2Institute for Bioscience and Biotechnology Interpretation of genomic variation plays an essential role in the analysis of Research, University of Maryland, Rockville, cancer and monogenic disease, and increasingly also in complex trait disease, with Maryland 3Department of Cell Biology and Molecular applications ranging from basic research to clinical decisions. Many computational Genetics, University of Maryland, College impact prediction methods have been developed, yet the field lacks a clear consensus Park, Maryland on their appropriate use and interpretation. The Critical Assessment of Genome 4Center for Computational Biology, University of California, Berkeley, California Interpretation (CAGI, /'kā‐jē/) is a community experiment to objectively assess computational methods for predicting the phenotypic impacts of genomic variation. Correspondence John Moult, Institute for Bioscience and CAGI participants are provided genetic variants and make blind predictions of Biotechnology Research, University of resulting phenotype. Independent assessors evaluate the predictions by comparing Maryland, 9600 Gudelsky Drive, Rockville, MD 20850. with experimental and clinical data. Email: [email protected] CAGI has completed five editions with the goals of establishing the state of art in Steven E. Brenner, Department of Plant and genome interpretation and of encouraging new methodological developments. This Microbial Biology, University of California, special issue (https://onlinelibrary.wiley.com/toc/10981004/2019/40/9) comprises Berkeley, CA 94720. -
The University of Chicago Interrogating the 3D Structure of Primate Genomes a Dissertation Submitted to the Faculty of the Divis
THE UNIVERSITY OF CHICAGO INTERROGATING THE 3D STRUCTURE OF PRIMATE GENOMES A DISSERTATION SUBMITTED TO THE FACULTY OF THE DIVISION OF THE BIOLOGICAL SCIENCES AND THE PRITZKER SCHOOL OF MEDICINE IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY DEPARTMENT OF HUMAN GENETICS BY ITTAI ETHAN ERES CHICAGO, ILLINOIS DECEMBER 2020 Copyright © 2020 by Ittai Ethan Eres All Rights Reserved Freely available under a CC-BY 4.0 International license \If I am not for myself, who will be for me? But if I am only for myself, who am I? And if not now, when?" Rabbi Hillel Table of Contents LIST OF FIGURES . vi LIST OF TABLES . vii ACKNOWLEDGMENTS . viii ABSTRACT . xi 1 INTRODUCTION . 1 1.1 The evolution of gene regulation . 1 1.2 Gene regulatory evolution insights from comparative primate genomics . 3 1.3 The growing importance of the 3D genome . 11 2 REORGANIZATION OF 3D GENOME STRUCTURE MAY CONTRIBUTE TO GENE REGULATORY EVOLUTION IN PRIMATES . 15 2.1 Abstract . 15 2.2 Introduction . 16 2.3 Results . 18 2.3.1 Inter-species differences in 3D genomic interactions . 19 2.3.2 The relationship between inter-species differences in contacts and gene expression . 26 2.3.3 The chromatin and epigenetic context of inter-species differences in 3D genome structure . 30 2.4 Discussion . 33 2.4.1 Contribution of variation in 3D genome structure to expression diver- gence . 36 2.4.2 Functional annotations . 37 2.5 Materials and methods . 38 2.5.1 Ethics statement . 38 2.5.2 Induced pluripotent stem cells (iPSCs) . -

Full Schedule ILANIT 20 -23 of February 2017
Full Schedule ILANIT 20th-23rd of February 2017 ILANIT / FISEB Federation of all the Israel Societies for Experimental Biology (FISEB) איגוד האגודות הישראליות לביולוגיה ניסויית )אילנית( ILANIT/FISEB is a federation of 31 Israeli societies of experimental biology. ILANIT’s conference is held every three years in Eilat, with attendance by researchers and students. This conference, held in February 2017, is the culmination of the most exciting research performed in Israel in a variety of biological disciplines. Board President Treasurer Secretary Karen B. Avraham Yaron Shav-Tal Eitan Yefenof (TAU) (BIU) (HUJI) Scientific Organizing Committee Conference President Conference Deputy Conference Vice Conference Vice Orna Amster-Choder President President President (HUJI) Angel Porgador (BGU) Maya Schuldiner (WIS) Eli Pikarsky (HUJI) 2 Last updated 16.02.2017 Scientific Advisory Committee Molecular and Structural Biology and Biochemistry Orna Elroy-Stein, Tel Aviv University (Chair) Ora Furman, Hebrew University of Jerusalem Shula Michaeli, Bar Ilan University Neurobiology and Endocrinology Yuval Dor, Hebrew University of Jerusalem (Chair) Yadin Dudai, Weizmann Institute of Science Assaf Rudich, Ben Gurion University of the Negev Genetics, Genomics, Epigenetics, Bioinformatics and Systems Biology Tzachi Pilpel, Weizmann Institute of Science (Chair) Ohad Birk, Ben Gurion University of the Negev Howard Cedar, Hebrew University of Jerusalem Yael Mandel, Technion – Israel Institute of Technology Medicine, Immunology and Cancer Roni Apte, -

Engineering Proteins from Sequence Statistics: Identifying and Understanding the Roles
Engineering Proteins from Sequence Statistics: Identifying and Understanding the Roles of Conservation and Correlation in Triosephosphate Isomerase DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Brandon Joseph Sullivan B.S. Graduate Program in the Ohio State Biochemistry Program The Ohio State University 2011 Dissertation Committee: Thomas J. Magliery, Advisor Mark P. Foster William C. Ray Copyright by Brandon Joseph Sullivan 2011 Abstract The structure, function and dynamics of proteins are determined by the physical and chemical properties of their amino acids. Unfortunately, the information encapsulated within a position or between positions is poorly understood. Multiple sequence alignments of protein families allow us to interrogate these questions statistically. Here, we describe the characterization of bioinformatically-designed variants of triosephosphate isomerase (TIM). First, we review the state-of-the-art for engineering proteins with increased stability. We examine two methodologies that benefit from the availability of large numbers - high-throughput screening and sequence statistics of protein families. Second, we have deconvoluted what properties are encoded within a position (conservation) and between positions (correlations) by designing TIMs in which each position is the most common amino acid in the multiple sequence alignment. We found that a consensus TIM from a raw sequence database performs the complex isomerization reaction with weak activity as a dynamic molten globule. Furthermore, we have confirmed that the monomeric species is the catalytically active conformation despite being designed from 600+ dimeric proteins. A second consensus TIM from a curated dataset is well folded, has wild-type activity and is dimeric, but it only differs from the raw consensus TIM at 35 nonconserved positions. -
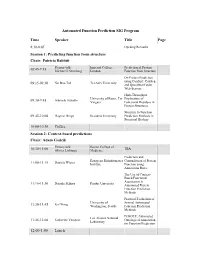
Automated Function Prediction SIG Program
Automated Function Prediction SIG Program Time Speaker Title Page 8:30-8:45 Opening Remarks Session 1: Predicting function from structure Chair: Patricia Babbitt Plenary talk: Imperial College, Prediction of Protein 08:45-9:15 Michael J. Sternberg London Function from Structure On Protein Prediction using ConSurf, ConSeq Nir Ben-Tal Tel Aviv University 09:15-09:30 and QuasiMotiFinder Web-Servers High-Throughput University of Rome, Tor Exploration of Gabriele Ausiello 09:30-9:45 Vergata Functional Residues in Protein Structures Structure to Function 09:45-10:00 Dagmar Ringe Brandeis University Prediction Methods in Structural Biology 10:00-10:30 Coffee Session 2: Context-based predictions Chair: Adam Godzik Plenary talk: Baylor College of TBA 10:30-11:00 Olivier Lichtarge Medicine Prediction and European Bioinformatics Contradiction of Protein Daniela Wieser 11:00-11:15 Institute Function using Annotation Rules The Use of Context- Based Functional Association in Daisuke Kihara Purdue University 11:15-11:30 Automated Protein Function Prediction Methods Practical Evaluation of University of Several Automated 11:30-11:45 Kai Wang Washington, Seattle Function Prediction Methods POSOLE: Automated Los Alamos National Katherine Verspoor Ontological Annotation 11:45-12:00 Laboratory for Function Prediction 12:00-1:00 Lunch Time Speaker Title Page Session 3: Analysis of functional sites Chair: Iddo Friedberg Plenary Talk: The FEATURE System 1:00-1:30 Stanford University for Protein Structure Russ Altman Annotation Structure-Based Function University -

Plos Computational Biology Publishes Research of Exceptional
A peer-reviewed open-access journal published by the Public Library of Science www.ploscompbiol.org PLoS Computational Biology publishes research of exceptional significance that furthers our understanding of living systems at all scales — from molecules and cells, to patient populations and ecosystems — through the application of computational methods. • Run by an international Editorial Board led by Philip E. Bourne (University of California San Diego, USA). • Featuring high-quality Research Articles, invited Reviews, Tutorials, an outstanding Education section, Image credit: Toma Pigli and our popular Editorial “Ten Simple Rules” series. PLoS Computational Biology (2007) • Funder-compliant — Published articles are Topics include (but are not limited to): immediately deposited in PubMed Central and Molecular Biology subsequently cited in PubMed. Protein-Protein Interactions • Provides constructive peer review and rapid Computational Neuroscience publication. Regulatory Networks • Article-level metrics and web tools to facilitate Computational Immunology community discourse through notes, comments, Sequence Analysis and ratings. Protein Structure & Function Prediction • Highlighted in news outlets and blogs from around Population Biology the world. Cancer Genetics Microarray Data Analysis Gene Expression Synthetic Biology PLoS Computational Biology is published by the Public Machine Learning Library of Science (PLoS), a nonprofit organization committed to making the world’s scientific and medical literature a public resource. Everything we publish is freely available online through- out the world, for anyone to read, download, copy, distribute and use (with attribution). Barrier-free, open access, no permissions required. Image credit: Ryan Davey PLoS Computational Biology (2007) PUBLIC LIBRARY of SCIENCE www.plos.org A peer-reviewed open-access journal published by the Public Library of Science www.ploscompbiol.org Editorial Board Philip E. -

Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by Pickeringhutchins Ltd
European Bioinformatics Institute EMBL-EBI Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by PickeringHutchins Ltd www.pickeringhutchins.com EMBL member states: Austria, Croatia, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, United Kingdom. Associate member state: Australia EMBL-EBI is a part of the European Molecular Biology Laboratory (EMBL) EMBL-EBI EMBL-EBI EMBL-EBI EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus, Hinxton Cambridge CB10 1SD United Kingdom Tel. +44 (0)1223 494 444, Fax +44 (0)1223 494 468 www.ebi.ac.uk EMBL Heidelberg Meyerhofstraße 1 69117 Heidelberg Germany Tel. +49 (0)6221 3870, Fax +49 (0)6221 387 8306 www.embl.org [email protected] EMBL Grenoble 6, rue Jules Horowitz, BP181 38042 Grenoble, Cedex 9 France Tel. +33 (0)476 20 7269, Fax +33 (0)476 20 2199 EMBL Hamburg c/o DESY Notkestraße 85 22603 Hamburg Germany Tel. +49 (0)4089 902 110, Fax +49 (0)4089 902 149 EMBL Monterotondo Adriano Buzzati-Traverso Campus Via Ramarini, 32 00015 Monterotondo (Rome) Italy Tel. +39 (0)6900 91402, Fax +39 (0)6900 91406 © 2012 EMBL-European Bioinformatics Institute All texts written by EBI-EMBL Group and Team Leaders. This publication was produced by the EBI’s Outreach and Training Programme. Contents Introduction Foreword 2 Major Achievements 2011 4 Services Rolf Apweiler and Ewan Birney: Protein and nucleotide data 10 Guy Cochrane: The European Nucleotide Archive 14 Paul Flicek: -

Annotation Inconsistencies Beyond Sequence Similarity-Based Function Prediction – Phylogeny and Genome Structure Vasilis J
Promponas et al. Standards in Genomic Sciences (2015) 10:108 DOI 10.1186/s40793-015-0101-2 COMMENTARY Open Access Annotation inconsistencies beyond sequence similarity-based function prediction – phylogeny and genome structure Vasilis J. Promponas1, Ioannis Iliopoulos2 and Christos A. Ouzounis3* Abstract The function annotation process in computational biology has increasingly shifted from the traditional characterization of individual biochemical roles of protein molecules to the system-wide detection of entire metabolic pathways and genomic structures. The so-called genome-aware methods broaden misannotation inconsistencies in genome sequences beyond protein function assignments, encompassing phylogenetic anomalies and artifactual genomic regions. We outline three categories of error propagation in databases by providing striking examples – at various levels of appreciation by the community from traditional to emerging, thus raising awareness for future solutions. Keywords: Genome-wide annotation, Genome-aware methods, Protein function prediction, Next-generation sequencing, Mis-annotation modeling, Error propagation, Genome structure, Genome evolution Background detected [6]. The strategy of sequence similarity-based Genome-wide sequence annotation typically involves the function prediction is well-understood, as the general cat- prediction of gene structure and regulation, protein egories of intrinsic errors for genome-wide annotation function and metabolic capabilities of a given species have been identified [7]. Previously, we have attempted to [1]. Despite (or perhaps because of) the high degree classify those categories into a quasi-quantitative scale of automation, genome annotation is an error-prone (named Transitive Annotation-Based Scale or TABS), in process [2]. As long as annotations are revisited and order to compare different annotation sets for a given curated on a continuous basis, risks associated with false genomic collection [1].