Photoactivatable Fluorophores
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Single-Molecule Spectroscopy and Imaging of Biomolecules in Living Cells
Anal. Chem. 2010, 82, 2192–2203 Single-Molecule Spectroscopy and Imaging of Biomolecules in Living Cells Samuel J. Lord, Hsiao-lu D. Lee, and W. E. Moerner* Department of Chemistry, Stanford University, Stanford, California 94305-5080 The number of reports per year on single-molecule imaging instance, the shape of the distribution may be skewed or reveal experiments has grown roughly exponentially since the first multiple subpopulations, which may offer insight into underlying successful efforts to optically detect a single molecule were mechanisms. Each single molecule is a local reporter on the completed over two decades ago. Single-molecule spectros- makeup and conditions of its immediate surroundings, its “na- copy has developed into a field that includes a wealth of noenvironment”, and thus acts as a readout of spatial heterogene- experiments at room temperature and inside living cells. The ity of a sample. SMS also measures time-dependent processes that fast growth of single-molecule biophysics has resulted from are not necessarily synchronized throughout the sample or its benefits in probing heterogeneous populations, one population. For example, multiple catalytic states of an enzyme molecule at a time, as well as from advances in microscopes will be convolved with all the states of other copies in an ensemble, and detectors. This Perspective summarizes the field of live- whereas a SMS experiment can measure uncorrelated stochastic cell imaging of single biomolecules. transitions of a single enzyme. SMS also has the ability to observe intermediate states or rare events, given that the instruments have Single-molecule biophysics spans a range of experiments, from sufficient time resolution. -

Rational Design of Small Molecule Fluorescent Probes for Biological Applications
Organic & Biomolecular Chemistry Rational Design of Small Molecule Fluorescent Probes for Biological Applications Journal: Organic & Biomolecular Chemistry Manuscript ID OB-REV-06-2020-001131.R1 Article Type: Review Article Date Submitted by the 13-Jul-2020 Author: Complete List of Authors: Jun, Joomyung; University of Pennsylvania, Chemistry; Massachusetts Institute of Technology, Chemistry Chenoweth, David; University of Pennsylvania, Department of Chemistry Petersson, E.; University of Pennsylvania, Chemistry Page 1 of 16 Organic & Biomolecular Chemistry ARTICLE Rational Design of Small Molecule Fluorescent Probes for Biological Applications a,b a a,c Received 00th January 20xx, Joomyung V. Jun, David M. Chenoweth* and E. James Petersson* Accepted 00th January 20xx Fluorescent small molecules are powerful tools for visualizing biological events, embodying an essential facet of chemical DOI: 10.1039/x0xx00000x biology. Since the discovery of the first organic fluorophore, quinine, in 1845, both synthetic and theoretical efforts have endeavored to “modulate” fluorescent compounds. An advantage of synthetic dyes is the ability to employ modern organic chemistry strategies to tailor chemical structures and thereby rationally tune photophysical properties and functionality of the fluorophore. This review explores general factors affecting fluorophore excitation and emission spectra, molar absorption, Stokes shift, and quantum efficiency; and provides guidelines for chemist to create novel probes. Structure- property relationships concerning the substituents are discussed in detail with examples for several dye families. Then, we present a survey of functional probes based on PeT, FRET, and environmental or photo-sensitivity, focusing on representative recent work in each category. We believe that a full understanding of dyes with diverse chemical moieties enables the rational design of probes for the precise interrogation of biochemical and biological phenomena. -

Product Information Sheet
AAT Bioquest®, Inc. Product Technical Information Sheet Last Updated July 2012 Classic Calcium Detection Reagents Calcium acts as a universal second messenger in a variety of cells. Numerous functions of all types of cells are regulated by Ca2+ to a greater or lesser degree, thus calcium measurement is critical for numerous biological investigations. Since the 1920s, scientists have attempted to measure Ca2+, but few were successful due to limited availability of Ca2+ probes. The first reliable measurement of Ca2+ was performed by Ridgway and Ashley by injecting the photoprotein aequorin into the giant muscle fiber of the barnacle. Subsequently, in the 1980s, Tsien and colleagues produced a variety of fluorescent indicators. Among them Indo-1, Fura-2, Fluo-3 and Rhod-2 have been the most valuable dyes for measuring Ca2+ with a fluorescence instrument. Fluorescent probes that show spectral responses upon binding to Ca2+ have enabled researchers to investigate changes in intracellular free Ca2+ concentrations by using fluorescence microscopy, flow cytometry, fluorescence spectroscopy and fluorescence microplate readers. Most of these fluorescent indicators are derivatives of BAPTA chelators that incorporate a PET system responsive to calcium. FLIPR® and FlexStation™ instruments of Molecular Devices Corp., FDSS of Hamamatsu Corp. and NOVOstar™ of BMG Technologies have enabled high throughput measurement of calcium for GPCR and ion channel research. There are quite a few factors that need to be considered when selecting a fluorescent Ca2+ indicator. Spectral Properties: For UV excitation, Indo-1 and Fura-2 are widely used. Fluo-3 is preferred for 488 nm excitation while Rhod-2 and X-rhod are used for red emissions. -

Recent Advances in Chemical Biology Using Benzophenones and Diazirines As Radical Precursors
molecules Review Recent Advances in Chemical Biology Using Benzophenones and Diazirines as Radical Precursors 1,2, , 1,2, Muhammad Murtaza Hassan * y and Olasunkanmi O. Olaoye y 1 Department of Chemical and Physical Sciences, University of Toronto Mississauga, 3359 Mississauga Road North, Mississauga, ON L5L 1C6, Canada; [email protected] 2 Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada * Correspondence: [email protected]; Tel.: +1-905-569-4588 These authors contributed equally to this work. y Academic Editor: Edward Lee-Ruff Received: 18 April 2020; Accepted: 9 May 2020; Published: 13 May 2020 Abstract: The use of light-activated chemical probes to study biological interactions was first discovered in the 1960s, and has since found many applications in studying diseases and gaining deeper insight into various cellular mechanisms involving protein–protein, protein–nucleic acid, protein–ligand (drug, probe), and protein–co-factor interactions, among others. This technique, often referred to as photoaffinity labelling, uses radical precursors that react almost instantaneously to yield spatial and temporal information about the nature of the interaction and the interacting partner(s). This review focuses on the recent advances in chemical biology in the use of benzophenones and diazirines, two of the most commonly known light-activatable radical precursors, with a focus on the last three years, and is intended to provide a solid understanding of their chemical and biological principles and their applications. Keywords: photoaffinity labelling; benzophenone; diazirine; radical precursors; interactome; SABRE; hyperpolarizing agents; crosslinking; photochemistry 1. Introduction The use of radicals or photoactivatable radical precursors has become ubiquitous in the fields of medicinal chemistry and chemical biology in the past three decades. -

Ca2+-Activated Kca3.1 Potassium Channels Contribute to the Slow
www.nature.com/scientificreports OPEN Ca2+‑activated KCa3.1 potassium channels contribute to the slow afterhyperpolarization in L5 neocortical pyramidal neurons M. V. Roshchin, V. N. Ierusalimsky, P. M. Balaban & E. S. Nikitin* Layer 5 neocortical pyramidal neurons are known to display slow Ca2+‑dependent afterhyperpolarization (sAHP) after bursts of spikes, which is similar to the sAHP in CA1 hippocampal cells. However, the mechanisms of sAHP in the neocortex remain poorly understood. Here, we identifed the Ca2+‑gated potassium KCa3.1 channels as contributors to sAHP in ER81‑positive neocortical pyramidal neurons. Moreover, our experiments strongly suggest that the relationship between sAHP and KCa3.1 channels in a feedback mechanism underlies the adaptation of the spiking frequency of layer 5 pyramidal neurons. We demonstrated the relationship between KCa3.1 channels and sAHP using several parallel methods: electrophysiology, pharmacology, immunohistochemistry, and photoactivatable probes. Our experiments demonstrated that ER81 immunofuorescence in layer 5 co‑localized with KCa3.1 immunofuorescence in the soma. Targeted Ca2+ uncaging confrmed two major features of KCa3.1 channels: preferential somatodendritic localization and Ca2+‑driven gating. In addition, both the sAHP and the slow Ca2+‑induced hyperpolarizing current were sensitive to TRAM‑ 34, a selective blocker of KCa3.1 channels. Te neocortex is the largest part of the cortex (~ 90% of the entire cortex in humans), and plays a crucial role in higher functions of the brain such as interpretation of sensory information, formation and storage of long-term memory, language usage, and control of voluntary movements. Pyramidal neurons play a key role in all cortical activities and constitute ~ 80% of all neocortical neurons. -

Advances in Engineering of Fluorescent Proteins And
Author's personal copy Available online at www.sciencedirect.com Advances in engineering of fluorescent proteins and photoactivatable proteins with red emission Kiryl D Piatkevich and Vladislav V Verkhusha Monomeric fluorescent proteins of different colors are widely energy transfer (FRET) approach to three and four colors used to study behavior and targeting of proteins in living cells. in a single cell [3]. Fluorescent proteins that irreversibly change their spectral properties in response to light irradiation of a specific The RFPs, whose chromophores are formed by induction wavelength, or photoactivate, have become increasingly with light, are known as the photoactivatable FPs (PA- popular to image intracellular dynamics and superresolution RFPs). Two different groups of PA-RFPs are presently protein localization. Until recently, however, no optimized being distinguished. Members of the first group exhibit monomeric red fluorescent proteins and red photoactivatable an irreversible photoconversion from the non-fluorescent proteins have been available. Furthermore, monomeric or green fluorescent state to the red fluorescent state. fluorescent proteins, which change emission from blue to red Members of the second group undergo reversible photo- simply with time, so-called fluorescent timers, were developed switching between the non-fluorescent and fluorescent to study protein age and turnover. Understanding of chemical states. Introduction of photoactivatable FPs into cell mechanisms of the chromophore maturation or biology greatly extended the spatio-temporal limits of photoactivation into a red form will further advance engineering in vivo biological dynamics [4] and have become useful of fluorescent timers and photoactivatable proteins with tools for the superresolution microscopy approaches such enhanced and novel properties. -

Calcium Detection Probes & Assay Kits
Calcium Detection Probes & Assay Kits 2016-2017 Cal-520™ Cal-590™ Fluo-8® AAT Bioquest® Advancing Assay & Test Technologies Our Mission AAT Bioquest® is committed to constantly meet or exceed its customer’s requirements by providing consistently high quality products and services, and by encouraging continuous improvements in its long-term and daily operations. Our core value is Innovation and Customer Satisfaction. Our Story AAT Bioquest®, Inc. (formerly ABD Bioquest, Inc.) develops, manufactures and markets bioanalytical research reagents and kits to life sciences research, diagnostic R&D and drug discovery. We specialize in photometric detections including absorption (color), fluorescence and luminescence technologies. The Company's superior products enable life science researchers to better under- stand biochemistry, immunology, cell biology and molecular biology. AAT Bioquest offers a rapidly expanding list of enabling products. Besides the standard catalog products, we also offer custom services to meet the distinct needs of each customer. Our current services include custom synthesis of biological detection probes, custom development of biochemical, cell-based and diagnostic assays and custom high throughput screening of drug discovery targets. It is my greatest pleasure to welcome you to AAT Bioquest. We greatly appreciate the constant support of our valuable customers. While we continue to rapidly expand, our core value remains the same: Innovation and Customer Satisfaction. We are committed to being the leading provider of novel biological detection solutions. We promise to extend these values to you during the course of our service and to continue to support you with our new products and services. It is our greatest honor to receive valuable feedbacks and suggestions from you so that we can better serve your projects. -
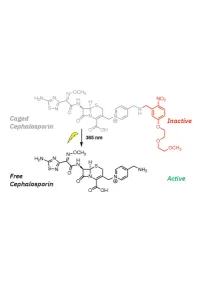
1 20 Introduction
1 Design, Synthesis, and Biological Evaluation of Light-Activated Antibiotics 2 3 Inga S. Shchelik, Andrea Tomio, and Karl Gademann 4 Department of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057, Zurich, SwitzerlanD 5 6 7 ABSTRACT 8 The spatial anD temporal control of bioactivity of small molecules by light (photopharmacology) constitutes a 9 promising approach for stuDy of biological processes and ultimately for the treatment of Diseases. In this stuDy, we 10 investigateD two Different ‘cageD’ antibiotic classes that can unDergo remote activation with UV-light at λ=365 nm, 11 via the conjugation of deactivating and photocleavable units through a short synthetic sequence. The two wiDely useD 12 antibiotics vancomycin and cephalosporin were thus enhanceD in their performance by renDering them 13 photoresponsive and thus suppressing undesired off-site activity. The antimicrobial activity against Bacillus subtilis 14 ATCC 6633, Staphylococcus aureus ATCC 29213, S. aureus ATCC 43300 (MRSA), Escherichia coli ATCC 25922, 15 and Pseudomonas aeruginosa ATCC 27853 could be spatiotemporally controlleD with light. Both molecular series 16 displayed a good activity winDow. The vancomycin Derivative DisplayeD excellent values against Gram-positive 17 strains after uncaging, and the next-generation caged cephalosporin derivative achieved good and broad activity 18 against both Gram-positive and Gram-negative strains after photorelease. 19 Key worDs: antibacterial agents, photopharmacology, photocaging, vancomycin, cephalosporin. 1 20 -

Physiological Probes & Assay Kits
Physiological Probes & Assay Kits Calcium Indicators · Membrane Potential Assays · pH Probes ® SIE HABEN DIE VISION, AAT Bioquest WIR HABEN DIE SUBSTANZ. Advancing Assay & Test Technologies www.aatbio.com Labeling Antibodies and Biopolymers Table of Contents Section 1 General Information 2 Section 6 Membrane Potential Measurement 33 Fast Response Membrane Potential Probes ................................................. 35 Section 2 Calcium Ion Detection 5 Slow Response Membrane Potential Probes ............................................... 36 Mitochondrial Membrane Potential Probes ................................................ 37 Fluo-8® Calcium Ion Indicators............................................................... ...... 8 FLIPR® Membrane Potential Assay Kits ....................................................... 38 2 Cal-520™ Calcium Ion Indicators .................................................................10 Rhod-4™ Calcium Ion Indicator ...................................................................11 Section 7 Index 39 Solutions Labeling Fluorescence Optimized BTC Calcium Ion Indicator ...........................................................................12 Fura-2 Calcium Ion Indicator .......................................................................12 Fura-8™ Calcium Ion Indicator ....................................................................12 Alphabetical Index .................................................................................. 40 Indo-1 Calcium Ion Indicator.......................................................................13 -

Specific Protein Labeling with Caged Fluorophores for Dual-Color Imaging and Super-Resolution Microscopy in Living Cells
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Specific protein labeling with caged fluorophores for dual-color imaging and super-resolution Cite this: Chem. Sci.,2017,8,559 microscopy in living cells† Sebastian Hauke,‡a Alexander von Appen,‡a Tooba Quidwai,b Jonas Riesb and Richard Wombacher*a We present new fluorophore-conjugates for dual-color photoactivation and super-resolution imaging inside live mammalian cells. These custom-designed, photo-caged Q-rhodamines and fluoresceins are cell-permeable, bright and localize specifically to intracellular targets. We utilized established orthogonal protein labeling strategies to precisely attach the photoactivatable fluorophores to proteins with subsequent activation of fluorescence by irradiation with UV light. That way, diffusive cytosolic proteins, histone proteins as well as filigree mitochondrial networks and focal adhesion proteins were visualized inside living cells. We applied the new photoactivatable probes in inverse fluorescence recovery after Creative Commons Attribution-NonCommercial 3.0 Unported Licence. photo-bleaching (iFRAP) experiments, gaining real-time access to protein dynamics from live biological Received 12th May 2016 settings with resolution in space and time. Finally, we used the caged Q-rhodamine for photo-activated Accepted 1st September 2016 localization microscopy (PALM) on both fixed and live mammalian cells, where the superior molecular DOI: 10.1039/c6sc02088g brightness and photo-stability directly resulted in improved localization precisions for different protein www.rsc.org/chemicalscience targets. Introduction photoactivation experiments inside living cells have been mainly restricted to the application of photo-modulatable FPs 11,12 This article is licensed under a For decades, photoactivation experiments, based on small so far. -

Enzyme-Activated Fluorogenic Probes for Live-Cell and in Vivo Imaging Wen Chyan and Ronald T
Reviews Cite This: ACS Chem. Biol. 2018, 13, 1810−1823 Enzyme-Activated Fluorogenic Probes for Live-Cell and in Vivo Imaging Wen Chyan and Ronald T. Raines* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States ABSTRACT: Fluorogenic probes, small-molecule sensors that unmask brilliant fluorescence upon exposure to specific stimuli, are powerful tools for chemical biology. Those probes that respond to enzymatic catalysis illuminate the complex dynamics of biological processes at a level of spatiotemporal detail and sensitivity unmatched by other techniques. Here, we review recent advances in enzyme-activated fluorogenic probes for biological imaging. We organize our survey by enzyme classification, with emphasis on fluorophore masking strategies, modes of enzymatic activation, and the breadth of current and future applications. Key challenges such as probe selectivity and spectroscopic requirements are described alongside therapeutic, diagnostic, and theranostic opportunities. luorogenic probes are latent fluorophores that reveal their ance of a probe, the fluorescence masking strategy governs its F signal in response to environmental changes, interactions enzymic target and responsiveness. Accordingly, we focus this with analytes, or specific chemical reactions.1 Fluorogenic review on enzyme-catalyzed unmasking strategies that have probes are prepared by chemically modulating the fluorescence been used for imaging in live cells and in vivo. We also of a parent fluorophore, rendering it nonfluorescent until constrain our survey to the past five years and abstain from activation by a specific triggering event. Because of their high extensive discussion of parent fluorophore chemistry and − sensitivity and ability to monitor diverse events selectively, spectroscopic properties covered elsewhere.3,16 19 fluorogenic probes are important components in the toolkit of 2−4 chemical biology. -

Novel Covalent Probes for Mapping Α7 Nicotinic Acetylcholine Receptor Allosteric Site/S
Novel Covalent Probes For Mapping α7 Nicotinic Acetylcholine Receptor Allosteric Site/s Master’s Thesis Research Defense by Vasantha Duggirala Advisor: Ganesh A. Thakur, Ph.D Department of Pharmaceutical Sciences Northeastern University November 2013 1 ACKNOWLEDGEMENTS I would like to express my deep appreciation to Dr. Ganesh Thakur for giving me a wonderful opportunity in his Drug Discovery lab and pursue Thesis under his guidance. I am grateful for the quality time he gave me to develop me as a person with character as well as teach science. I am obliged to sit through his medicinal chemistry classes for the strong concepts and realize real time drug discovery research process which I would not have had if I chose to graduate by just doing coursework. I thank him for having confidence in me and boosting my morale when the project was under stake. He is one teacher who not only wishes his students should develop into learned professionals but also into amazing individuals with self-esteem and personality. I would like to thank Dr. Purnima Mungalachetty for her willingness to be my Thesis committee member and spare her precious time out of a very busy schedule to review and guide me through Thesis. I am inspired by her dedication and perseverance to excel in this field and be a leader ultimately. I am very pleased to acknowledge Dr. John S. Gatley, my academic advisor and well-wisher from the bottom of my heart for his confidence in me and to his valuable criticism which helped me give my Thesis a good shape.