5817 Ani Con 2.1 Ward
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bantengbanteng Populationpopulation Inin Cambodia:Cambodia: Thethe Establishedestablished Baselinebaseline Densitydensity © FA / WWF-Cambodia
FACTSHEET 2011 BantengBanteng PopulationPopulation inin Cambodia:Cambodia: TheThe EstablishedEstablished BaselineBaseline DensityDensity © FA / WWF-Cambodia Between 2009-2011 in dry seasons, the research team of WWF-Cambodia conducted the first vigorous surveys on population abundance of large mammals which includes wild cattle, deer, and wild pig in the Eastern Plain Landscape (EPL) of Cambodia covering an area of approximately 6,000km2. Banteng: Globally Endangered Species Banteng (bos javanicus) is a species of wild cattle that historically inhabited deciduous and semi- evergreen forests from Northeast India and Southern Yunnan through mainland Southeast Asia and Peninsular Malaysia to Borneo and Java. Since 1996, banteng has been listed by IUCN as globally endangered on the basis of an inferred decline over the last 30 years of more than 50%. Banteng is most likely the ancestor of Southeast Asia’s domestic cattle and it is considered to be one of the most beautiful and graceful of all wild cattle species. In Cambodia, banteng populations have decreased dramatically since the late 1960s. Poaching to sell the meat and horns as trophies constitutes a major threat to remnant populations even though banteng is legally protected. © FA / WWF-Cambodia Monitoring Banteng Population in the Landscape Knowledge of animal populations is central to understanding their status and to planning their management and conservation. That is why WWF has several research projects in the EPL to gain more information about the biodiversity values of PPWS and MPF. Regular line transect surveys are conducted to collect data on large ungulates like banteng, gaur, and Eld’s deer--all potential prey species for large carnivores including tigers. -

The “Big Five” on Land &
58-25 Queens Blvd. Woodside, NY 11377 T: (718) 280-5000; (800) 627-1244 F: (718) 204-4726 E: [email protected] W: www.classicescapes.com Nature & Cultural Journeys for the Discerning Traveler YOU ARE CORDIALLY INVITED TO JOIN THE BROOKFIELD ZOO IN COOPERATION WITH THE SHEDD AQUARIUM ON A WILDLIFE & MARINE ADVENTURE TO SOUTH AFRICA THE “BIG FIVE” ON LAND & SEA NOVEMBER 3 TO 15, 2019 . Schedules, accommodations and prices are accurate at the time of writing. They are subject to change YOUR ITINERARY DAY 1 ~ SUNDAY ~ NOVEMBER 3 CHICAGO / EN ROUTE Your adventure begins as you board your overnight flight to Johannesburg. (Meals Aloft) DAY 2 ~ MONDAY ~ NOVEMBER 4 CAPE TOWN This afternoon, arrive in Johannesburg where you connect with your flight to Cape Town, South Africa’s “Mother City”. Upon arrival, you will be met by your specialist guide and escorted to your hotel. The provincial capital, Cape Town, is a sophisticated city with plenty to see and do, particularly around the Victoria and Alfred Waterfront area, where delightful buildings of the Cape Dutch and Victorian-era architecture have been restored as shops, restaurants, museums and pubs, while the busy water traffic of the docks goes on unabated. Your home for the next three nights, the Vineyard hotel and Spa, with over 200 years of history within its walls, this deluxe hotel is situated in six acres of attractive landscaped parkland on the banks of the Liesbeeck River. Located in the lush leafy suburb of Newlands, the Vineyard Hotel & Spa is within easy walking distance of the up-market Cavendish Shopping Centre and is just 10 minutes away from the City Center and the popular Victoria & Alfred Waterfront. -

Bison Literature Review Biology
Bison Literature Review Ben Baldwin and Kody Menghini The purpose of this document is to compare the biology, ecology and basic behavior of cattle and bison for a management context. The literature related to bison is extensive and broad in scope covering the full continuum of domestication. The information incorporated in this review is focused on bison in more or less “wild” or free-ranging situations i.e.., not bison in close confinement or commercial production. While the scientific literature provides a solid basis for much of the basic biology and ecology, there is a wealth of information related to management implications and guidelines that is not captured. Much of the current information related to bison management, behavior (especially social organization) and practical knowledge is available through local experts, current research that has yet to be published, or popular literature. These sources, while harder to find and usually more localized in scope, provide crucial information pertaining to bison management. Biology Diet Composition Bison evolutional history provides the basis for many of the differences between bison and cattle. Bison due to their evolution in North America ecosystems are better adapted than introduced cattle, especially in grass dominated systems such as prairies. Many of these areas historically had relatively low quality forage. Bison are capable of more efficient digestion of low-quality forage then cattle (Peden et al. 1973; Plumb and Dodd 1993). Peden et al. (1973) also found that bison could consume greater quantities of low protein and poor quality forage then cattle. Bison and cattle have significant dietary overlap, but there are slight differences as well. -

Buffalo Hunt: International Trade and the Virtual Extinction of the North American Bison
NBER WORKING PAPER SERIES BUFFALO HUNT: INTERNATIONAL TRADE AND THE VIRTUAL EXTINCTION OF THE NORTH AMERICAN BISON M. Scott Taylor Working Paper 12969 http://www.nber.org/papers/w12969 NATIONAL BUREAU OF ECONOMIC RESEARCH 1050 Massachusetts Avenue Cambridge, MA 02138 March 2007 I am grateful to seminar participants at the University of British Columbia, the University of Calgary, the Environmental Economics workshop at the NBER Summer Institute 2006, the fall 2006 meetings of the NBER ITI group, and participants at the SURED II conference in Ascona Switzerland. Thanks also to Chris Auld, Ed Barbier, John Boyce, Ann Carlos, Charlie Kolstad, Herb Emery, Mukesh Eswaran, Francisco Gonzalez, Keith Head, Frank Lewis, Mike McKee, and Sjak Smulders for comments; to Michael Ferrantino for access to the International Trade Commission's library; and to Margarita Gres, Amanda McKee, Jeffrey Swartz, Judy Hasse of Buffalo Horn Ranch and Andy Strangeman of Investra Ltd. for research assistance. Funding for this research was provided by the SSHRC. The views expressed herein are those of the author(s) and do not necessarily reflect the views of the National Bureau of Economic Research. © 2007 by M. Scott Taylor. All rights reserved. Short sections of text, not to exceed two paragraphs, may be quoted without explicit permission provided that full credit, including © notice, is given to the source. Buffalo Hunt: International Trade and the Virtual Extinction of the North American Bison M. Scott Taylor NBER Working Paper No. 12969 March 2007 JEL No. F1,Q2,Q5,Q56 ABSTRACT In the 16th century, North America contained 25-30 million buffalo; by the late 19th century less than 100 remained. -

Conservation Status of Asiatic Wild Buffalo (Bubalus Arnee) in Chhattisgarh
Conservation status of Asiatic Wild Buffalo (Bubalus arnee) in Chhattisgarh revealed through genetic study A Technical Report Prepared by Laboratory for the Conservation of Wildlife Trust of India Endangered Species(LACONES) F – 13, Sector 08 CSIR-CCMB Annex I, HYDERABAD – 500048 NCR, Noida - 201301 Disclaimer: This publication is meant for authorized use by laboratories and persons involved in research on conservation of Wild buffalos. LaCONES shall not be liable for any direct, consequential or incidental damages arising out of the protocols described in this book. Reference to any specific product (commercial or non-commercial), processes or services by brand or trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation or favor by LaCONES. The information and statements contained in this document shall not be used for the purpose of advertising or to imply the endorsement or recommendation of LaCONES. Citation: Mishra R.P. and A. Gaur. 2019. Conservation status of Asiatic Wild Buffalo (Bubalus arnee) in Chhattisgarh revealed through genetic study. Technical Report of WTI and CSIR-CCMB, 17p ACKNOWLEDGEMENTS We are thankful to the Forest Department, Govt. of Chhattisgarh for giving permission to carry out the conservation and research activities on Wild buffalo in various protected areas in Chhattisgarh. We are grateful to Shri Ram Prakash, PCCF (Retd.); Shri R.N. Mishra, PCCF (Retd.); Dr. R.K. Singh, PCCF (Retd.), Shri Atul Kumar Shukla, Principal Chief Conservator of Forests & Chief Wildlife Warden and Dr. S.K. Singh, Additional Principal Chief Conservator of Forests (WL), Dr. Rakesh Mishra, Director CSIR-CCMB, Dr. Rahul Kaul, Executive Director, WTI, Dr. -

Infectious Diseases of Saiga Antelopes and Domestic Livestock in Kazakhstan
Infectious diseases of saiga antelopes and domestic livestock in Kazakhstan Monica Lundervold University of Warwick, UK June 2001 1 Chapter 1 Introduction This thesis combines an investigation of the ecology of a wild ungulate, the saiga antelope (Saiga tatarica, Pallas), with epidemiological work on the diseases that this species shares with domestic livestock. The main focus is on foot-and-mouth disease (FMD) and brucellosis. The area of study was Kazakhstan (located in Central Asia, Figure 1.1), home to the largest population of saiga antelope in the world (Bekenov et al., 1998). Kazakhstan's independence from the Soviet Union in 1991 led to a dramatic economic decline, accompanied by a massive reduction in livestock numbers and a virtual collapse in veterinary services (Goskomstat, 1996; Morin, 1998a). As the rural economy has disintegrated, the saiga has suffered a dramatic increase in poaching (Bekenov et al., 1998). Thus the investigation reported in this thesis includes ecological, epidemiological and socio-economic aspects, all of which were necessary in order to gain a full picture of the dynamics of the infectious diseases of saigas and livestock in Kazakhstan. The saiga is an interesting species to study because it is one of the few wildlife populations in the world that has been successfully managed for commercial hunting over a period of more than 40 years (Milner-Gulland, 1994a). Its location in Central Asia, an area that was completely closed to foreigners during the Soviet era, means that very little information on the species and its management has been available in western literature. The diseases that saigas share with domestic livestock have been a particular focus of this study because of the interesting issues related to veterinary care and disease control in the Former Soviet Union (FSU). -

Wild Yak Bos Mutus in Nepal: Rediscovery of a Flagship Species
Mammalia 2015; aop Raju Acharya, Yadav Ghimirey*, Geraldine Werhahn, Naresh Kusi, Bidhan Adhikary and Binod Kunwar Wild yak Bos mutus in Nepal: rediscovery of a flagship species DOI 10.1515/mammalia-2015-0066 2009). It has also been believed to inhabit the lower eleva- Received April 15, 2015; accepted July 21, 2015 tion Altai ranges in Mongolia (Olsen 1990). The possible fossil of wild yak discovered in Nepal (Olsen 1990) provided historical evidence of the species’ Abstract: Wild yak Bos mutus is believed to have gone presence in the country. Schaller and Liu (1996) also extinct from Nepal. Various searches in the last decade stated the occurrence of the wild yak in Nepal. Wild failed to document its presence. In Humla district, far- yak is said to inhabit the areas north of the Himalayas western Nepal, we used observation from transects and (Jerdon 1874, Hinton and Fry 1923), which also include vantage points, sign survey on trails, and informal dis- Limi Valley. Skins and horns were sporadically discov- cussions to ascertain the presence of wild yak in 2013 ered by explorers in the Himalayan and trans-Himala- (May–June) and 2014 (June–July). Direct sightings of two yan region of the country. These include evidence of individuals and hoof marks, dung piles, pelts, and head three horns of the species (around four decades old) of an individual killed in 2012 confirmed its presence. that can still be found in Lo Manthang (two pairs of The wild yak has an uncertain national status with con- horn) and Tsaile (one pair of horns) of Upper Mustang firmed records only from Humla district. -

Prowling for Predators- Africa Overnight
Prowling for Predators- Africa Overnight: SCHEDULE: 6:45- 7:00 Arrive 7:00- 8:20 Introductions Zoo Rules Itinerary Introduction to Predator/Prey dynamics- presented with live animal encounters Food Pyramid Talk 8:20- 8:45 Snack 8:45-11:00 Building Tours 11:00-11:30 HOPE Jeopardy PREPARATION: x Paint QUESTing spots with blacklight Paint x Hide clue tubes NEEDS: x Zoo Maps x Charged Blacklight Flashlights (Triple As) x Animal Food Chain Cards x Ball of String x Hula Hoops, Tablecloths ANIMAL OPTIONS: x Ball Python x Hedgehog x Tarantula x Flamingos x Hornbill x White-Faced Scops Owl x Barn Owl x Radiated Tortoise x Spiny-Tailed Lizard DEPENDING ON YOUR ORDER YOU WILL: Tour Buildings: x Commissary- QUESTing o Front: Kitchen o Back: Dry Foods x AFRICA o Front: African QUESTing- Lion o Back: African QUESTing- Cheetah x Reptile House- QUESTing o King Cobra (Right of building) Animal Demos: x In the Education Building Games: x Africa Outpost I **manageable group sizes in auditorium or classrooms x Oh Antelope x Quick Frozen Critters x HOPE Jeopardy x Africa Outpost II o HOPE Jeopardy o *Overflow game: Musk Ox Maneuvers INTRODUCTION & HIKE INFORMATION (AGE GROUP SPECIFIC) x See appendix I Prowling for Predators: Africa Outpost I Time Requirement: 4hrs. Group Size & Grades: Up to 100 people- 2nd-4t h grades Materials: QUESTing handouts Goals: -Create a sense of WONDER to all participants -We can capitalize on wonder- During up-close animal demos & in front of exhibit animals/behind the scenes opportunities. -Convey KNOWLEDGE to all participants -This should be done by using participatory teaching methods (e.g. -
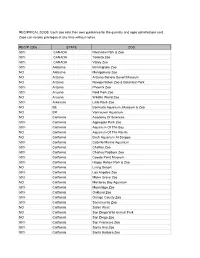
2006 Reciprocal List
RECIPRICAL ZOOS. Each zoo sets their own guidelines for the quantity and ages admitted per card. Zoos can revoke privileges at any time without notice. RECIP 2006 STATE ZOO 50% CANADA Riverview Park & Zoo 50% CANADA Toronto Zoo 50% CANADA Valley Zoo 50% Alabama Birmingham Zoo NO Alabama Montgomery Zoo NO Arizona Arizona-Sonora Desert Museum NO Arizona Navajo Nation Zoo & Botanical Park 50% Arizona Phoenix Zoo 50% Arizona Reid Park Zoo NO Arizona Wildlife World Zoo 50% Arkansas Little Rock Zoo NO BE Bermuda Aquarium, Museum & Zoo NO BR Vancouver Aquarium NO California Academy Of Sciences 50% California Applegate Park Zoo 50% California Aquarium Of The Bay NO California Aquarium Of The Pacific NO California Birch Aquarium At Scripps 50% California Cabrillo Marine Aquarium 50% California Chaffee Zoo 50% California Charles Paddock Zoo 50% California Coyote Point Museum 50% California Happy Hollow Park & Zoo NO California Living Desert 50% California Los Angeles Zoo 50% California Micke Grove Zoo NO California Monterey Bay Aquarium 50% California Moonridge Zoo 50% California Oakland Zoo 50% California Orange County Zoo 50% California Sacramento Zoo NO California Safari West NO California San Diego Wild Animal Park NO California San Diego Zoo 50% California San Francisco Zoo 50% California Santa Ana Zoo 50% California Santa Barbara Zoo NO California Seaworld San Diego 50% California Sequoia Park Zoo NO California Six Flags Marine World NO California Steinhart Aquarium NO CANADA Calgary Zoo 50% Colorado Butterfly Pavilion NO Colorado Cheyenne -

Species of the Day: Banteng
Images © Brent Huffman / Ultimate Ungulate © Brent Huffman Species of the Day: Banteng The Banteng, Bos javanicus, is listed as ‘Endangered’ on the IUCN Red List of Threatened SpeciesTM. A species of wild cattle, the Banteng occurs in Southeast Asia from Myanmar to Indonesia, with a large introduced population in northern Australia. The Banteng has been eradicated from much of its historical range, and the remaining wild population, estimated at no more than 8,000 individuals, is continuing to decline. Habitat loss Geographical range and hunting present the greatest threats to its survival, with the illegal trade in meat and horns www.iucnredlist.org still being widespread in Southeast Asia. www.asianwildcattle.org Help Save Species Although the Banteng is legally protected across its range and occurs in a number of www.arkive.org protected areas, the natural resources of reserves in Southeast Asia often continue to be exploited. The northern Australian population may offer a conservation alternative, although genetic studies hint that the stock may originate from domesticated Bali cattle. Fortunately, a captive population is maintained worldwide which, if managed effectively and supplemented occasionally, can provide a buffer against total extinction, and offer the potential for future re- introductions into the wild. The production of the IUCN Red List of Threatened Species™ is made possible through the IUCN Red List Partnership: Species of the Day IUCN (including the Species Survival Commission), BirdLife is sponsored by International, Conservation International, NatureServe and Zoological Society of London.. -

The Friday Edition September 29 2017 Home Advantage
PROPERTYINSIDE: 34-PAGESPECIAL HOME ADVANTAGE THE FRIDAY EDITION SEPTEMBER 29 2017 FE80_Cover_PRESS.indd 1 11/09/2017 16:53 THE SHARPENER alpaca punch Strong yet soft, smart yet relaxed – it’s no wonder alpaca is leading the pack this season, says Tom Stubbs fabric that’s extra light, versatile, strong yet utterly luxurious: it sounds like a menswear designer’s fantasy. But alpaca has, of course, been around for ages – it’s just that its superlative qualities have not beenA fully appreciated until this season. The springy, ultra-soft fibres from the underbellies and necks of a species of camelid living in the Andes make for some very special fabrics. When woven, alpaca takes on various textures, from soft and voluminous to coarse and cropped. And as lightweight fabrications and distinctive textures become defining characteristics of contemporary men’s style, it’s not surprising that alpaca is now being shepherded into a lead role. Brunello Cucinelli, who built his empire on cashmere, has also put alpaca to work beautifully in his signature unstructured tailored outerwear, such as a glen-check short coat (£3,760) and roomy one-and-a-half breasted camel- (£1,390) and bomber jackets (£1,060, colour coat (£3,890). Likewise at Canali, pictured below) in wool/alpaca/mohair/ where deconstructed drapey overcoats silk bouclé take inspiration from 1960s silhouettes, as does a single-breasted overcoat (£1,470) in a wool/alpaca blend. They pass muster at smart occasions, yet their subtle texture and soft construction mean they also work as weekend throw- ons. The highlight at Chester Barrie is a Change coat (£2,950, pictured below right), its navy cashmere contrasting Alpaca is ideal cable-knit turtleneck (£395) have with a lush black alpaca lapel (made by for upgrading a 1940s quality about them. -

Bison, Water Buffalo, &
February 2021 - cdfa' Bison, Water Buffalo, & Yak (or Crossbreeds) Entry Requirements ~ EPAlTMENT OF CALI FORNI \1c U LTU RE FOOD & AC Interstate Livestock Entry Permit California requires an Interstate Livestock Entry Permit for all bison, water buffalo, and/or yaks. To obtain an Interstate Livestock Entry Permit, please call the CDFA Animal Health Branch (AHB) permit line at (916) 900-5052. Permits are valid for 15 days after being issued. Certificate of Veterinary Inspection California requires a Certificate of Veterinary Inspection (CVI) for bison, water buffalo, and/or yaks within 30 days before movement into the state. Official Identification (ID) Bison, water buffalo, and/or yaks of any age and sex require official identification. Brucellosis Brucellosis vaccination is not required for bison, ------1Animal Health Branch Permit Line: water buffalo, and/or yaks to enter California. (916) 900-5052 A negative brucellosis test within 30 days prior to entry is required for all bison, water buffalo, and/ If you are transporting livestock into California or yaks 6 months of age and over with the with an electronic CVI, please print and present following exceptions: a hard copy to the Inspector at the Border • Steers or identified spayed heifers, and Protection Station. • Any Bovidae from a Certified Free Herd with the herd number and date of current Animal Health and Food Safety Services test recorded on the CVI. Animal Health Branch Headquarters - (916) 900-5002 Tuberculosis (TB) Redding District - (530) 225-2140 Modesto District - (209) 491-9350 A negative TB test is Tulare District - (559) 685-3500 required for all bison, Ontario District - (909) 947-4462 water buffalo, and/or yaks 6 months of age and over within For California entry requirements of other live- www.cdfa.ca.gov stock and animals, please visit the following: 60 days prior to Information About Livestock and Pet Movement movement.