UNIVERSITY of PÉCS Characterization of the Vestibular NADPH
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Imm Catalog.Pdf
$ Gene Symbol A B 3 C 4 D 9 E 10 F 11 G 12 H 13 I 14 J. K 17 L 18 M 19 N 20 O. P 22 R 26 S 27 T 30 U 32 V. W. X. Y. Z 33 A ® ® Gene Symbol Gene ID Antibody Monoclonal Antibody Polyclonal MaxPab Full-length Protein Partial-length Protein Antibody Pair KIt siRNA/Chimera Gene Symbol Gene ID Antibody Monoclonal Antibody Polyclonal MaxPab Full-length Protein Partial-length Protein Antibody Pair KIt siRNA/Chimera A1CF 29974 ● ● ADAMTS13 11093 ● ● ● ● ● A2M 2 ● ● ● ● ● ● ADAMTS20 80070 ● AACS 65985 ● ● ● ADAMTS5 11096 ● ● ● AANAT 15 ● ● ADAMTS8 11095 ● ● ● ● AATF 26574 ● ● ● ● ● ADAMTSL2 9719 ● AATK 9625 ● ● ● ● ADAMTSL4 54507 ● ● ABCA1 19 ● ● ● ● ● ADAR 103 ● ● ABCA5 23461 ● ● ADARB1 104 ● ● ● ● ABCA7 10347 ● ADARB2 105 ● ABCB9 23457 ● ● ● ● ● ADAT1 23536 ● ● ABCC4 10257 ● ● ● ● ADAT2 134637 ● ● ABCC5 10057 ● ● ● ● ● ADAT3 113179 ● ● ● ABCC8 6833 ● ● ● ● ADCY10 55811 ● ● ABCD2 225 ● ADD1 118 ● ● ● ● ● ● ABCD4 5826 ● ● ● ADD3 120 ● ● ● ABCG1 9619 ● ● ● ● ● ADH5 128 ● ● ● ● ● ● ABL1 25 ● ● ADIPOQ 9370 ● ● ● ● ● ABL2 27 ● ● ● ● ● ADK 132 ● ● ● ● ● ABO 28 ● ● ADM 133 ● ● ● ABP1 26 ● ● ● ● ● ADNP 23394 ● ● ● ● ABR 29 ● ● ● ● ● ADORA1 134 ● ● ACAA2 10449 ● ● ● ● ADORA2A 135 ● ● ● ● ● ● ● ACAN 176 ● ● ● ● ● ● ADORA2B 136 ● ● ACE 1636 ● ● ● ● ADRA1A 148 ● ● ● ● ACE2 59272 ● ● ADRA1B 147 ● ● ACER2 340485 ● ADRA2A 150 ● ● ACHE 43 ● ● ● ● ● ● ADRB1 153 ● ● ACIN1 22985 ● ● ● ADRB2 154 ● ● ● ● ● ACOX1 51 ● ● ● ● ● ADRB3 155 ● ● ● ● ACP5 54 ● ● ● ● ● ● ● ADRBK1 156 ● ● ● ● ACSF2 80221 ● ● ADRM1 11047 ● ● ● ● ACSF3 197322 ● ● AEBP1 165 ● ● ● ● ACSL4 2182 ● -

Identification of SH3 Domain Proteins Interacting with the Cytoplasmic Tail of the a Disintegrin and Metalloprotease 10 (ADAM10)
Identification of SH3 Domain Proteins Interacting with the Cytoplasmic Tail of the A Disintegrin and Metalloprotease 10 (ADAM10) Henriette Ebsen, Marcus Lettau, Dieter Kabelitz, Ottmar Janssen* University of Kiel, Molecular Immunology, Institute for Immunology, University Hospital Schleswig-Holstein Campus Kiel, Kiel, Germany Abstract The a disintegrin and metalloproteases (ADAMs) play a pivotal role in the control of development, adhesion, migration, inflammation and cancer. Although numerous substrates of ADAM10 have been identified, the regulation of its surface expression and proteolytic activity is still poorly defined. One current hypothesis is that both processes are in part modulated by protein-protein interactions mediated by the intracellular portion of the protease. For related proteases, especially proline-rich regions serving as docking sites for Src homology domain 3 (SH3) domain-containing proteins proved to be important for mediating regulatory interactions. In order to identify ADAM10-binding SH3 domain proteins, we screened the All SH3 Domain Phager library comprising 305 human SH3 domains using a GST fusion protein with the intracellular region of human ADAM10 as a bait for selection. Of a total of 291 analyzed phage clones, we found 38 SH3 domains that were precipitated with the ADAM10-derived fusion protein but not with GST. We verified the binding to the cytosolic portion of ADAM10 for several candidates by co-immunoprecipitation and/or pull down analyses. Intriguingly, several of the identified proteins have been implicated in regulating surface appearance and/or proteolytic activity of related ADAMs. Thus, it seems likely that they also play a role in ADAM10 biology. Citation: Ebsen H, Lettau M, Kabelitz D, Janssen O (2014) Identification of SH3 Domain Proteins Interacting with the Cytoplasmic Tail of the A Disintegrin and Metalloprotease 10 (ADAM10). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

GAK and PRKCD Are Positive Regulators of PRKN-Independent
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.05.369496; this version posted November 5, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 GAK and PRKCD are positive regulators of PRKN-independent 2 mitophagy 3 Michael J. Munson1,2*, Benan J. Mathai1,2, Laura Trachsel1,2, Matthew Yoke Wui Ng1,2, Laura 4 Rodriguez de la Ballina1,2, Sebastian W. Schultz2,3, Yahyah Aman4, Alf H. Lystad1,2, Sakshi 5 Singh1,2, Sachin Singh 2,3, Jørgen Wesche2,3, Evandro F. Fang4, Anne Simonsen1,2* 6 1Division of Biochemistry, Department of Molecular Medicine, Institute of Basic Medical Sciences, University of Oslo 7 2Centre for Cancer Cell Reprogramming, Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, N-0316, Oslo, Norway. 8 3Department of Molecular Cell Biology, The Norwegian Radium Hospital Montebello, N-0379, Oslo, Norway 9 4Department of Clinical Molecular Biology, University of Oslo and Akershus University Hospital, 1478 Lørenskog, Norway 10 11 Keywords: GAK, Cyclin G Associated Kinase, PRKCD, Protein Kinase C Delta, Mitophagy, DFP, 12 DMOG, PRKN 13 14 *Corresponding Authors: 15 [email protected] 16 [email protected] 17 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.05.369496; this version posted November 5, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Identification of Genomic Targets of Krüppel-Like Factor 9 in Mouse Hippocampal
Identification of Genomic Targets of Krüppel-like Factor 9 in Mouse Hippocampal Neurons: Evidence for a role in modulating peripheral circadian clocks by Joseph R. Knoedler A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Neuroscience) in the University of Michigan 2016 Doctoral Committee: Professor Robert J. Denver, Chair Professor Daniel Goldman Professor Diane Robins Professor Audrey Seasholtz Associate Professor Bing Ye ©Joseph R. Knoedler All Rights Reserved 2016 To my parents, who never once questioned my decision to become the other kind of doctor, And to Lucy, who has pushed me to be a better person from day one. ii Acknowledgements I have a huge number of people to thank for having made it to this point, so in no particular order: -I would like to thank my adviser, Dr. Robert J. Denver, for his guidance, encouragement, and patience over the last seven years; his mentorship has been indispensable for my growth as a scientist -I would also like to thank my committee members, Drs. Audrey Seasholtz, Dan Goldman, Diane Robins and Bing Ye, for their constructive feedback and their willingness to meet in a frequently cold, windowless room across campus from where they work -I am hugely indebted to Pia Bagamasbad and Yasuhiro Kyono for teaching me almost everything I know about molecular biology and bioinformatics, and to Arasakumar Subramani for his tireless work during the home stretch to my dissertation -I am grateful for the Neuroscience Program leadership and staff, in particular -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Flavone Effects on the Proteome and Transcriptome of Colonocytes in Vitro and in Vivo and Its Relevance for Cancer Prevention and Therapy
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährungsphysiologie Flavone effects on the proteome and transcriptome of colonocytes in vitro and in vivo and its relevance for cancer prevention and therapy Isabel Winkelmann Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. D. Haller Prüfer der Dissertation: 1. Univ.-Prof. Dr. H. Daniel 2. Univ.-Prof. Dr. U. Wenzel (Justus-Liebig-Universität Giessen) 3. Prof. Dr. E.C.M. Mariman (Maastricht University, Niederlande) schriftliche Beurteilung Die Dissertation wurde am 24.08.2009 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 25.11.2009 angenommen. Die Forschung ist immer auf dem Wege, nie am Ziel. (Adolf Pichler) Table of contents 1. Introduction .......................................................................................................... 1 1.1. Cancer and carcinogenesis .................................................................................. 2 1.2. Colorectal Cancer ............................................................................................... 3 1.2.1. Hereditary forms of CRC ........................................................................................ 4 1.2.2. Sporadic forms of CRC .......................................................................................... -

Downloaded from Ensembl
UCSF UC San Francisco Electronic Theses and Dissertations Title Detecting genetic similarity between complex human traits by exploring their common molecular mechanism Permalink https://escholarship.org/uc/item/1k40s443 Author Gu, Jialiang Publication Date 2019 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California by Submitted in partial satisfaction of the requirements for degree of in in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO AND UNIVERSITY OF CALIFORNIA, BERKELEY Approved: ______________________________________________________________________________ Chair ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ Committee Members ii Acknowledgement This project would not have been possible without Prof. Dr. Hao Li, Dr. Jiashun Zheng and Dr. Chris Fuller at the University of California, San Francisco (UCSF) and Caribou Bioscience. The Li lab grew into a multi-facet research group consist of both experimentalists and computational biologists covering three research areas including cellular/molecular mechanism of ageing, genetic determinants of complex human traits and structure, function, evolution of gene regulatory network. Labs like these are the pillar of global success and reputation -

Identification of Genomic Alterations in Castration Resistant Prostate Cancer Using Next Generation Sequencing
Identification of Genomic Alterations in Castration Resistant Prostate Cancer using Next Generation Sequencing Thesis Submitted for a Doctoral Degree in Natural Sciences (Dr. rer. nat) Faculty of Mathematics and Natural Sciences Rheinische Friedrich-Wilhelms- Submitted by Roopika Menon from Chandigarh, India Bonn 2013 Prepared with the consent of the Faculty of Mathematics and Natural Sciences at the Rheinische Friedrich-Wilhelms- 1. Reviewer: Prof. Dr. Sven Perner 2. Reviewer: Prof. Dr. Hubert Schorle Date of examination: 19 November 2013 Year of Publication: 2014 Declaration I solemnly declare that the work submitted here is the result of my own investigation, except where otherwise stated. This work has not been submitted to any other University or Institute towards the partial fulfillment of any degree. ____________________________________________________________________ Roopika Menon; Author Acknowledgements This thesis would not have been possible without the help and support of many people. I would like to dedicate this thesis to all the people who have helped make this dream a reality. This thesis would have not been possible without the patience, support and guidance of my supervisor, Prof. Dr. Sven Perner. It has truly been an honor to be his first PhD student. He has both consciously and unconsciously made me into the researcher that I am today. My PhD experience has truly been the ‘best’ because of his time, ideas, funding and most importantly his incredible sense of humor. He encouraged and gave me the opportunity to travel around the world to develop as a scientist. I cannot thank him enough for this immense opportunity, which stands as a stepping-stone to my career in science. -

Identi Cation of Speci C Role of SNX Family in Gastric Cancer Prognosis
Identication of Specic Role of SNX Family in Gastric Cancer Prognosis Evaluation Beibei Hu First Aliated Hospital of China Medical University Guohui Yin Heibei University of Technology Xuren Sun ( [email protected] ) First Aliated Hospital of China Medical University Research Article Keywords: SNX family, gastric cancer, prognosis, bioinformatics, articial neural network Posted Date: August 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-832476/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/24 Abstract Project: We here perform a systematic bioinformatic analysis to uncover the role of SNX family in clinical outcome of gastric cancer (GC). Methods: Comprehensive bioinformatic analysis were realized with online tools such as TCGA, GEO, String, Timer, cBioportal and Kaplan-Meier Plotter. Statistic analysis was conducted with R language, and articial neural network was constructed using Python. Results: Our analysis demonstrated that SNX4/5/6/7/8/10/13/14/15/16/20/22/25/27/30 were higher expressed in GC, whereas SNX1/17/21/24/33 were in the opposite expression proles. Clustering results gave the relative transcriptional levels of 30 SNXs in tumor, and it was totally consistent to the inner relevance of SNXs at mRNA level. Protein-Protein Interaction (PPI) map showed closely and complex connection among 33 SNXs. Tumor immune inltration analysis asserted that SNX1/3/9/18/19/21/29/33, SNX1/17/18/20/21/29/31/33, SNX1/2/3/6/10/18/29/33, and SNX1/2/6/10/17/18/20/29 were strongly correlated with four kinds of survival related TIICs, including Cancer associated broblast, endothelial cells, macrophages and Tregs. -
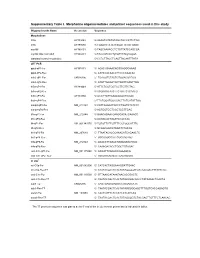
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Supplementary Figure 1. Epigenomic Maps of Endocrine Therapy Sensitive and Resistant Cell Lines
Supplementary Figure 1. Epigenomic maps of endocrine therapy sensitive and resistant cell lines. (A-C) ChIP-qPCR against H3K36me3 (A), H3K4me2 (B) or FAIRE-qPCR (C) was used to validate enrichment at randomly selected sites identified by ChIP-seq specific to MCF7 (left-side panel) or LTED (right-side panel) cells. Color code represents relative fold enrichment as calculated against an internal negative control. (D) Confocal analysis of immunofluorescence assay against ERα protein in MCF7-LTED HA (>12 mo) cells. Primary (prim.) and secondary (sec.) antibody controls are shown. (E) GSC analysis of the overlap between two previously published ERα cistromes and the epigenomic maps obtained in this study. Supplementary Figure 2. ERα prognostic target genes are coherently expressed in endocrine therapy resistant cell lines. (A) RT-qPCR analysis of ERα target genes across endocrine therapy-responsive or - resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and SEM of replicates are shown. (B) RT-qPCR analysis of ERα expression across endocrine therapy-responsive or -resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and standard error of the mean (SEM) of replicates are shown. Supplementary Figure 3. Reprogramming of the chromatin cascade increases Notch activity in endocrine therapy resistant cell lines. (A-B) RT-qPCR analysis of two γ-secretase complex subunits across endocrine therapy- responsive or -resistant cell lines. Expression levels are normalized against MCF7 cells (dotted line). The average and SEM of replicates are shown. (C-D-E) Kaplan-Meier plots of Luminal A, Luminal B or ET patients using Notch3 expression probe.