A TEM Investigation of Metamict Samarskite from 1 the Garnet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And Ta-Bearing Oxide Minerals in the Greer Lake Pegmatitic Granite and Its Pegmatiteaureole, Southeasternmanitoba
American Mineralogist, Volume 71, pages 501-517, 1986 Fractionation trends of the Nb- and Ta-bearing oxide minerals in the Greer Lake pegmatitic granite and its pegmatiteaureole, southeasternManitoba Pnrn ennmi, Bnucn E. Golo,r FuNx C. H,lwruronNr, RoN CrHplr,lN Department of Earth Sciences,University of Manitoba, Winnipeg, Manitoba R3T 2N2, Canada Arsrucr The Greer Lake pegmatitic granite and related exterior rare-element pegmatites of the beryl-columbite type intrude metabasalt and tonalite gneiss in the Archean English River Subprovince of southeasternManitoba. Columbite-tantalite is the predominant Nb- and Ta-bearingmineral, associatedwith subordinateixiolite, microlite, niobian-tantalian rutile, and rare tantalian cassiterite, wodginite, and ilmenite. In coexisting mineral pairs, Tal(Ta + Nb) of microlite exceedsthat of columbite-tantalite, ixiolite, rutile, and cassiterite;in cassiteriteand rutile, Tal(Ta + Nb) is higher and Mn/(Mn * Fe) lower than in columbite- tantalite and ixiolite. In Li-, Rb-, Cs-, and F-poor environments, limited Mn enrichment accompaniesthe fractionation of Ta, which culminates in ixiolite and subordinate microlite. In Li-, Rb-, Cs-, and F-rich parageneses,extensive Mn enrichment precedesthe main Ta fractionation, which subsequentlygenerates near-end-member manganotantalite, wodginite, and mi- crolite. The Greer Lake and other fractionation trends indicate that a late-stageF-rich environment promotes extreme Fe/Mn fractionation prior to the main stage of Nb-Ta separation. The abundancesof Sn, Ti, and Sc are not related to fluorine or rare-alkali enrichment, but increasefrom pegmatitic granite to pegnatites. The relative accumulation of Ti and Sc in the pegmatite aureole seemsto be due to internal fractionation rather than assimilation. Columbite-tantalite in the pegmatitic granite shows an intermediate to near-ordered structural state, but is highly disordered in the pegmatite veins. -

Niobian Rutile from the Mcguire Granitic Pegmatite, Park County, Colorado: Solid Solution, Exsolution, and Oxidation
American Mineralogist, Volume 84, pages 754–763, 1999 Niobian rutile from the McGuire granitic pegmatite, Park County, Colorado: Solid solution, exsolution, and oxidation PETR Cˇ ERNY´ ,1,* RON CHAPMAN,1 WILLIAM B. SIMMONS,2 AND LEONARD E. CHACKOWSKY1 1 Department of Geological Sciences, University of Manitoba, Winnipeg, Manitoba R3T 2N2, Canada 2 Department of Geology and Geophysics, University of New Orleans, New Orleans, Louisiana 70148-2850, U.S.A. ABSTRACT Coarse crystals of niobian rutile occur in the hydrothermally altered core-margin zone of the McGuire granitic pegmatite, Park County, Colorado, associated with potassium feldspar, quartz, bi- otite, ilmenite, and monazite-(Ce). Primary homogeneous niobian rutile, with Fe3+ @ Fe2+ and a small excess of (Fe,Mn) over the amount required to compensate the incorporation of (Nb,Ta,W), under- went three stages of exsolution. Primary homogeneous niobian rutile exsolved a fine trellis-like pat- tern of minor lamellar Nb-bearing pseudorutile I. Most of this phase was broken down to pseudomor- phs consisting of microgranular Nb-rich pseudorutile II imbedded in niobian-ferrian “ferropseudobrookite.” Continued exsolution in niobian rutile and reconstitution of the early exsolution products generated (Fe,Nb)-depleted, microgranular niobian rutile, titanian ferrocolumbite, and mi- nor ilmenite. These three phases did not attain chemical equilibrium but may represent a stable phase assemblage. All these processes seem to have maintained charge balance, suggesting a closed sys- tem. Subsequent to the three stages of exsolution, extensive oxidation converted the mineral assem- blages to anatase + hematite + titanian-tungstenian ixiolite; primary ilmenite was oxidized into an anatase + hematite intergrowth. In both cases, the hematite component was almost completely leached out, leaving highly porous aggregates of the other phases. -

The Crystal Chemistry of the Tapiolite Series
631 The Canadian M ineralo g is t Vol. 34, pp.631,-647(1,996) THE CRYSTALCHEMISTRY OFTHE TAPIOLITE SERIES MICHAELA. WISE Departnunt of Mincral Sciences,Snr.ithsonian hstitution" Washington,D,C, 20560, U.SA. PETR dERNf Depamnentof GeologicalSciences, Unitersity of Maninba. Winnipeg,Manitoba R3T2N2 ABsrRAc"r Ferotapiolite-manganotapiolite-groupminslzls [@e,Mn)Ta2O6]axe formed as :rccessoryphases in rare-elementgranitic pegmatitesexhibiting moderateto high degreesof fractionation.Despite their relative paucity, 94 setsof unit-cell parameters and 194 chemical compositionswere compiled to characterizethe crystal chemistry of the tapiolite series.X-ray-diffraction studiesindicate that the degreeof orderand compositional variations stongly influenceunit-cell dimensionsof tapiolite.Natural tapiolite showswide rangesof structuralstate. Crystallization of disorderedphases at low lemperaturesis suggested;however, the available data are not unambiguous.Tapiolite chemistry is typically uniform (Fe > Mn, Ta > Nb), and compositional variations are the result of limited, but effective homovalentard heterovalentsubstitutions: (1) Mn2+Fe2+-r;(2) NbsiTas+-l; (3) Fek(ti,Sn)4rFe2+_r(IaM)5*-r and (4) @,sn)4,1Fe,Mn;{,6M,Ta)5*_2. ln association witl otler M,Ta,Sn-bearing minerals,tapiolite shows a distinct preferencefor Fd+ and Ta over Mn and Nb. Emicbment in Nb, Ti and Sn appearsto be commonin tapiolite from moderatelyfractionated pegmatites, whereas extreme Mn enrichmentis typical of highly fractionated pegmatitesof the petalite subtypeor metasomaticunits. Keywords:tapiolite series,crystal chemistry,order - disorder,tantalum" granitic pegmatite. SoM:uaRs Irs mindraux de la s6de ferrotapiolite - manganotapiolitetGe"IUn)TqO6l se pr6sententsous forme d'accessoiresdes pegmatitesgranitiques d 6l6mentsrares faisantpreuve d'un degr6de fractionnementintermddiaire tr 61ev6.Malgrd leur raret6, les r6sultatsde 94 affinementsde la maille 6l6mentaireet de 194 analyseschimiques sont disponibles,et serventi caract6riser la cristallochimie des min6raux de cette s6rie. -

Tantalum Mineralogy, Rare-Element Granitic Pegmatites, Separation Lake
Ontario Geological Survey Open File Report 6022 Tantalum Mineralogy of Rare-Element Granitic Pegmatites from the Separation Lake Area, Northwestern Ontario 2000 ONTARIO GEOLOGICAL SURVEY Open File Report 6022 Tantalum Mineralogy of Rare-Element Granitic Pegmatites from the Separation Lake Area, Northwestern Ontario by A.G. Tindle and F.W. Breaks 2000 Parts of this publication may be quoted if credit is given. It is recommended that reference to this publication be made in the following form: Tindle, A.G. and Breaks, F.W. 2000. Tantalum mineralogy of rare-element granitic pegmatites from the Separation Lake area, northwestern Ontario; Ontario Geological Survey, Open File Report 6022, 378p. e Queen’s Printer for Ontario, 2000 e Queen’s Printer for Ontario, 2000. Open File Reports of the Ontario Geological Survey are available for viewing at the Mines Library in Sudbury, at the Mines and Minerals Information Centre in Toronto, and at the regional Mines and Minerals office whose district includes the area covered by the report (see below). Copies can be purchased at Publication Sales and the office whose district includes the area covered by the report. Al- though a particular report may not be in stock at locations other than the Publication Sales office in Sudbury, they can generally be obtained within 3 working days. All telephone, fax, mail and e-mail orders should be directed to the Publica- tion Sales office in Sudbury. Use of VISA or MasterCard ensures the fastest possible service. Cheques or money orders should be made payable to the Minister of Finance. Mines and Minerals Information Centre (MMIC) Tel: (416) 314-3800 Macdonald Block, Room M2-17 1-800-665-4480(toll free inside Ontario) 900 Bay St. -
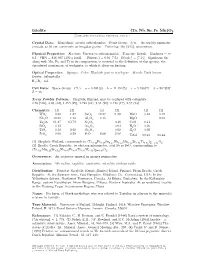
Ixiolite (Ta, Nb, Sn, Fe, Mn)O2 C 2001-2005 Mineral Data Publishing, Version 1
Ixiolite (Ta, Nb, Sn, Fe, Mn)O2 c 2001-2005 Mineral Data Publishing, version 1 Crystal Data: Monoclinic, pseudo-orthorhombic. Point Group: 2/m. As crudely prismatic crystals, to 10 cm; commonly as irregular grains. Twinning: On {013}, uncommon. Physical Properties: Fracture: Uneven to subconchoidal. Tenacity: Brittle. Hardness = 6– 6.5 VHN = 836–967 (100 g load). D(meas.) = 6.94–7.23 D(calc.) = [7.34] Significant Sn, along with Mn, Fe, and Ti in its composition, is essential to the definition of this species, the disordered counterpart of wodginite, to which it alters on heating. Optical Properties: Opaque. Color: Blackish gray to steel-gray. Streak: Dark brown. Luster: Submetallic. R1–R2: n.d. Cell Data: Space Group: P 2/c. a = 9.481(3) b = 11.494(5) c = 5.158(2) β =90◦8(2)0 Z=16 X-ray Powder Pattern: Skogb¨ole,Finland; may be confused with columbite. 2.98 (100), 3.65 (32), 1.459 (29), 1.722 (24), 2.51 (20), 1.746 (17), 2.57 (13) Chemistry: (1) (2) (1) (2) (1) (2) WO3 0.30 1.87 SnO2 12.27 11.38 MnO 5.40 9.19 Nb2O5 10.50 6.12 Al2O3 0.16 MgO 0.01 Ta2O5 61.47 63.79 Sc2O3 0.16 CaO 0.11 + SiO2 0.12 As2O3 0.04 H2O 0.16 − TiO2 0.38 2.68 Sb2O3 0.02 H2O 0.08 ZrO2 0.60 0.20 FeO 8.08 2.98 Total 99.63 98.44 (1) Skogb¨ole,Finland; corresponds to (Ta0.44Fe0.18Sn0.13Nb0.12Mn0.12Zr0.01Ti0.01)Σ=1.01O2. -

Primary Minerals of the Jáchymov Ore District
Journal of the Czech Geological Society 48/34(2003) 19 Primary minerals of the Jáchymov ore district Primární minerály jáchymovského rudního revíru (237 figs, 160 tabs) PETR ONDRU1 FRANTIEK VESELOVSKÝ1 ANANDA GABAOVÁ1 JAN HLOUEK2 VLADIMÍR REIN3 IVAN VAVØÍN1 ROMAN SKÁLA1 JIØÍ SEJKORA4 MILAN DRÁBEK1 1 Czech Geological Survey, Klárov 3, CZ-118 21 Prague 1 2 U Roháèových kasáren 24, CZ-100 00 Prague 10 3 Institute of Rock Structure and Mechanics, V Holeovièkách 41, CZ-182 09, Prague 8 4 National Museum, Václavské námìstí 68, CZ-115 79, Prague 1 One hundred and seventeen primary mineral species are described and/or referenced. Approximately seventy primary minerals were known from the district before the present study. All known reliable data on the individual minerals from Jáchymov are presented. New and more complete X-ray powder diffraction data for argentopyrite, sternbergite, and an unusual (Co,Fe)-rammelsbergite are presented. The follow- ing chapters describe some unknown minerals, erroneously quoted minerals and imperfectly identified minerals. The present work increases the number of all identified, described and/or referenced minerals in the Jáchymov ore district to 384. Key words: primary minerals, XRD, microprobe, unit-cell parameters, Jáchymov. History of mineralogical research of the Jáchymov Chemical analyses ore district Polished sections were first studied under the micro- A systematic study of Jáchymov minerals commenced scope for the identification of minerals and definition early after World War II, during the period of 19471950. of their relations. Suitable sections were selected for This work was aimed at supporting uranium exploitation. electron microprobe (EMP) study and analyses, and in- However, due to the general political situation and the teresting domains were marked. -

Cation Distribution in Partially Ordered Columbite from the Kings Mountain
American Mineralogist, Volume 76, pages 1897-1904, 1991 Cation distribution in partially ordered columbite from the Kings Mountain pegmatite,North Carolina M,qnc WnNGrn.TrroNr.ts Anrvrnnusrnn I-aboratorium fiir chemischeund mineralogischeKristallographie, Universitit Bern, Freiestrasse3, CH-3012 Bern, Switzerland Cnlllns A. Gnrcnn BayerischesForschungsinstitut {iir experimentelle Geochemie und Geophysik, Universitdt Bayreuth, Postfach IOl25l, D-8580 Bayreuth, Germany ABSTRAcT Two columbite samples,space group Pbcn, with a : 14.233,b : 5.724,c : 5.104 A, Z: 4 and a: 14.221,b : 5.727,c : 5.102A, of similar chemistry(average composition FeourMno.roTi005Nb.TaorOu)but from different rock types of the Kings Mountain peg- matite, North Carolina, were investigated. One columbite specimen (NCP5) originates from a primary spodumene pegmatite, the other (NCPI) from a rock composed mainly of sugaryalbite formed by subsequenthydrothermal activity. Single-crystalX-ray structure refinements indicate that both columbite samplesshow strong disorder between (Nb,Ta) and (Fe,Mn) on octahedral A and B positions. This is in agreementwith a Mdssbauer spectrum (NCP5), which was fitted with two doublets characteristicof two distinct t61Fe2+ sites. Both samples show strong chemical zoning with respect to Nb and Ta, but weak zoning of Fe and Mn. The similar chemical composition and cation ordering indicate that columbite in both rocks is formed by the same process.Late hydrothermal alteration of the original spodumenepegmatite did not affect the composition of the earlier crystallized columbite. The primary columbite (NCP5) shows slightly stronger disorder of two- and five-valent cations than columbite (NCPI) found in the hydrothermally altered part of the pegmatite. INrnonucrroN some samplesreveal a wodginite pattern (Khvostova and Maximova, 1969),whereas others yield a columbite pat- Minerals of the columbite-tantalite group have the gen- tern (Nickel et al., 1963b). -

Mineralogy and Petrology of the Amazonite Pegmatite at Bakstevalåsen, Øvre Eiker
Master Thesis, Department of Geosciences Mineralogy and petrology of the amazonite pegmatite at Bakstevalåsen, øvre Eiker Øyvind Sunde Mineralogy and petrology of the amazonite pegmatite at Bakstevalåsen, øvre Eiker Øyvind Sunde Master Thesis in Geosciences Discipline: Geology Department of Geosciences Faculty of Mathematics and Natural Sciences University of Oslo July 2013 © Øyvind Sunde, 2013 Supervised by associate prof. Rune S. Selbekk and prof. Tom Andersen Cover picture: Hand specimen of the amazonite pegmatite at Bakstevalåsen measuring a 15 cm cross-section with amazonite matrix and abundant danalite. This work is published digitally through DUO – Digitale Utgivelser ved UiO http://www.duo.uio.no It is also catalogued in BIBSYS (http://www.bibsys.no/english) All rights reserved. No part of this publication may be reproduced or transmitted, in any form or by any means, without permission. Acknowledgements This thesis marks the end of a 5 –year period of time with relentless studies at the Department of Geosciences, University of Oslo. There are many people I have met during this 5-year ride who in various ways have contributed in shaping my interest for geology. I have never, ever, regretted my decision on setting sail onto this journey. You all know who you are and a huge thank you! My thesis would not have been possible without the help of several clever individuals, and I would like to aim a special appreciation to the following personnel: Rune Selbekk: first of all, thank you for letting me volunteer at the natural History Museum during my infant years of studying. It brought more geology into a curriculum diluted with meteorology and philosophy. -

Geochemistry of Cassiterite and Wolframite from Tin and Tungsten Quartz Veins in Portugal ⁎ A.M.R
Available online at www.sciencedirect.com Ore Geology Reviews 33 (2008) 221–238 www.elsevier.com/locate/oregeorev Geochemistry of cassiterite and wolframite from tin and tungsten quartz veins in Portugal ⁎ A.M.R. Neiva Department of Earth Sciences, University of Coimbra, 3000-272 Coimbra, Portugal Received 9 August 2005; accepted 29 May 2006 Available online 4 October 2007 Abstract Cassiterite and wolframite compositions from SnNW- and WNSn-bearing quartz veins in Northern and Central Portugal are compared to provide evidence on fluid compositions. In SnNW-bearing quartz veins, euhedral cassiterite shows sequences of alternating parallel darker and lighter zones. The darker zones are pleochroic, oscillatory zoned, exhibit exsolutions of columbite and ixiolite and are richer in Nb, Ta and Fe than the lighter zones which consist of nearly pure SnO2. Cassiterite from WNSn-bearing quartz veins is usually zoned, with homogeneous and slightly pleochroic darker zones, which are chemically similar to lighter zones. Both zones have inclusions of rutile and rare ilmenite. The darker zones of cassiterite from the former veins are richer in Nb, Ta and Fe contents and poorer in Ti than the darker and lighter zones of cassiterite from the latter veins. This is attributed to differences in the composition of magmatic hydrothermal fluids. Wolframite compositions from SnNW- and W NSn-bearing quartz veins do not show any significant distinction, because they precipitate from relatively similar magmatic hydrothermal fluids. In some deposits, most wolframite crystals are homogeneous, but others are heterogeneous. Inner patches, rich in a hübnerite component, rarely occur in crystals from the Filharoso and Panasqueira deposits. -

The Wodginite Group. I. Structural Crystallography
597 Canadian Mineralogist Vol. 30, pp. 597-611 (1992) THE WODGINITE GROUP. I. STRUCTURAL CRYSTALLOGRAPHY T. SCOTT ERCIT Mineral Sciences Section, Canadian Museum of Nature, Ottawa, Ontario KIP 6P4 FRANK C. HAWTHORNE AND PETR CERNY Department of Geological Sciences, University of Ma nitoba, Winnipeg, Ma nitoba R3T 2N2 ABSTRACT Four new crystal-structure refinements have been done on wodginite-group minerals. Two partially ordered samples were used in single-crystal experiments, one, tantalian, the other, ferroan. The ferroan sample was heated to induce order, and its structure was re-refined. In addition to the single-crystal experiments, a Rietveld refinement from powder-diffraction data was done on a fully ordered lithiowodginite. Refinements of the partially ordered samples show that they are structurally intermediate to wodginite and ixiolite, and that heating under conventional conditions of 1000°C for 16 hours induces full wodginite-type cation order. In terms of both individual and mean bond-lengths, data for Li-rich samples deviate significantly from established trends for Li-poorer samples in a manner consistent with disorder of Li off the central A position. Schemes of cation order for potential derivatives of ixiolite have been evaluated for cells up to four times the volume of the ixiolite cell = size of the wodginite cell). For full ABC208 cation order, four most-probable schemes of order result. Three of these involve a doubled cell; one involves a quadrupled cell, relative to an ixiolite parent. Only the scheme of order with a quadrupled cell occurs naturally, i. e. , the wodginite structure; there is nothing in its scheme of order to indicate why nature prefers it over the other three schemes; more sophisticated calculations of energy seem necessary. -

Geochemistry of Niobium and Tantalum
Geochemistry of Niobium and Tantalum GEOLOGICAL SURVEY PROFESSIONAL PAPER 612 Geochemistry of Niobium and Tantalum By RAYMOND L. PARKER and MICHAEL FLEISCHER GEOLOGICAL SURVEY PROFESSIONAL PAPER 612 A review of the geochemistry of niobium and tantalum and a glossary of niobium and tantalum minerals UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1968 UNITED STATES DEPARTMENT OF THE INTERIOR STEWART L. UDALL, Secretary GEOLOGICAL SURVEY William T. Pecora, Director Library of Congress catalog-card No. GS 68-344 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 50 cents (paper cover) CONTENTS Page Page Abstract_ _ __-_.. _____________________ 1 Geochemical behavior Continued Introduction. _________________________ 2 Magmatic rocks Continued General geochemical considerations. _____ 2 Volcanic rock series______--____---__.__-_-__ 2. Abundance of niobium and tantalum_____ 3 Sedimentary rocks______________________________ 2. Crustal abundance-________________ 3 Deposits of niobium and tantalum.___________________ 2£ Limitations of data________________ 3 Suggestions for future work__--___-_------__-___---_- 26 Abundance in rocks._______________ 5 References, exclusive of glossary______________________ 27 Qualifying statement.__________ 5 Glossary of niobium and tantalum minerals.___________ 3C Igneous rocks_________________ 6 Part I Classification of minerals of niobium and Sedimentary rocks.____________ 10 tantalum according to chemical types_________ 31 Abundance in meteorites and tektites. 12 Part II Niobium and tantalum minerals..-_______ 32 Isomorphous substitution.______________ 13 Part III Minerals reported to contain 1-5 percent Geochemical behavior._________________ 15 niobium and tantalum_______________________ 38 Magma tic rocks ___________________ 15 Part IV Minerals in which niobium and tantalum Granitic rocks_________________ 16 have been detected in quantities less than 1 Albitized and greisenized granitic rocks. -

Amazonite: Mineralogy, Crystal Chemistry, and Typomorphism
Amazonite: Mineralogy, Crystal Chemistry, and Typomorphism Mikhail Ostrooumov Institute of Earth Sciences, University of Michoacan of San Nicolas of Hidalgo, Morelia, Mexico AMSTERDAM • BOSTON • HEIDELBERG • LONDON • NEW YORK • OXFORD PARIS • SAN DIEGO • SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO Elsevier Radarweg 29, PO Box 211, 1000 AE Amsterdam, Netherlands The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, UK 225 Wyman Street, Waltham, MA 02451, USA Copyright © 2016 Elsevier Inc. All rights reserved. This English language edition is a translation of original Russian language edition titled Амазонский камень, ISBN 978-5-7325-0675-4 Copyright notice: © Издательство “Политехника“, 2008. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or any information storage and retrieval system, without permission in writing from the publisher. Details on how to seek permission, further information about the Publisher’s permissions policies and our arrangements with organizations such as the Copyright Clearance Center and the Copyright Licensing Agency, can be found at our website: www.elsevier.com/permissions. This book and the individual contributions contained in it are protected under copyright by the Publisher (other than as may be noted herein). Notices Knowledge and best practice in this field are constantly changing. As new research and experience broaden our understanding, changes in research methods, professional practices, or medical treatment may become necessary. Practitioners and researchers must always rely on their own experience and kno wledge in evaluating and using any information, methods, compounds, or experiments described herein. In using such information or methods they should be mindful of their own safety and the safety of others, including parties for whom they have a professional responsibility.