Deep Mixed Model for Marginal Epistasis Detection and Population
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Gene-Level Methylome-Wide Association Analysis Identifies Novel
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201376; this version posted July 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 A gene-level methylome-wide association analysis identifies novel 2 Alzheimer’s disease genes 1 1 2 3 4 3 Chong Wu , Jonathan Bradley , Yanming Li , Lang Wu , and Hong-Wen Deng 1 4 Department of Statistics, Florida State University; 2 5 Department of Biostatistics & Data Science, University of Kansas Medical Center; 3 6 Population Sciences in the Pacific Program, University of Hawaii Cancer center; 4 7 Tulane Center for Biomedical Informatics and Genomics, Deming Department of Medicine, 8 Tulane University School of Medicine 9 Corresponding to: Chong Wu, Assistant Professor, Department of Statistics, Florida State 10 University, email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201376; this version posted July 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 11 Abstract 12 Motivation: Transcriptome-wide association studies (TWAS) have successfully facilitated the dis- 13 covery of novel genetic risk loci for many complex traits, including late-onset Alzheimer’s disease 14 (AD). However, most existing TWAS methods rely only on gene expression and ignore epige- 15 netic modification (i.e., DNA methylation) and functional regulatory information (i.e., enhancer- 16 promoter interactions), both of which contribute significantly to the genetic basis ofAD. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

TMEM59 (NM 004872) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP305624 TMEM59 (NM_004872) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human transmembrane protein 59 (TMEM59) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 36 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_004863 Locus ID: 9528 UniProt ID: Q9BXS4 RefSeq Size: 1709 Cytogenetics: 1p32.3 RefSeq ORF: 972 Synonyms: C1orf8; DCF1; HSPC001; PRO195; UNQ169 Summary: This gene encodes a protein shown to regulate autophagy in response to bacterial infection. This protein may also regulate the retention of amyloid precursor protein (APP) in the Golgi apparatus through its control of APP glycosylation. Overexpression of this protein has been found to promote apoptosis in a glioma cell line. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Feb 2015] Protein Families: Transmembrane This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 TMEM59 (NM_004872) Human Recombinant Protein – TP305624 Product images: Coomassie blue staining of purified TMEM59 protein (Cat# TP305624). -

Dynamic Transcriptomic Profiles of Zebrafish Gills in Response to Zinc
Zheng et al. BMC Genomics 2010, 11:548 http://www.biomedcentral.com/1471-2164/11/548 RESEARCH ARTICLE Open Access Dynamic transcriptomic profiles of zebrafish gills in response to zinc depletion Dongling Zheng1,4, Peter Kille2, Graham P Feeney2, Phil Cunningham1, Richard D Handy3, Christer Hogstrand1* Abstract Background: Zinc deficiency is detrimental to organisms, highlighting its role as an essential micronutrient contributing to numerous biological processes. To investigate the underlying molecular events invoked by zinc depletion we performed a temporal analysis of transcriptome changes observed within the zebrafish gill. This tissue represents a model system for studying ion absorption across polarised epithelial cells as it provides a major pathway for fish to acquire zinc directly from water whilst sharing a conserved zinc transporting system with mammals. Results: Zebrafish were treated with either zinc-depleted (water = 2.61 μgL-1; diet = 26 mg kg-1) or zinc-adequate (water = 16.3 μgL-1; diet = 233 mg kg-1) conditions for two weeks. Gill samples were collected at five time points and transcriptome changes analysed in quintuplicate using a 16K oligonucleotide array. Of the genes represented the expression of a total of 333 transcripts showed differential regulation by zinc depletion (having a fold-change greater than 1.8 and an adjusted P-value less than 0.1, controlling for a 10% False Discovery Rate). Down-regulation was dominant at most time points and distinct sets of genes were regulated at different stages. Annotation enrichment analysis revealed that ‘Developmental Process’ was the most significantly overrepresented Biological Process GO term (P = 0.0006), involving 26% of all regulated genes. -

The Interactome of KRAB Zinc Finger Proteins Reveals the Evolutionary History of Their Functional Diversification
Resource The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification Pierre-Yves Helleboid1,†, Moritz Heusel2,†, Julien Duc1, Cécile Piot1, Christian W Thorball1, Andrea Coluccio1, Julien Pontis1, Michaël Imbeault1, Priscilla Turelli1, Ruedi Aebersold2,3,* & Didier Trono1,** Abstract years ago (MYA) (Imbeault et al, 2017). Their products harbor an N-terminal KRAB (Kru¨ppel-associated box) domain related to that of Krüppel-associated box (KRAB)-containing zinc finger proteins Meisetz (a.k.a. PRDM9), a protein that originated prior to the diver- (KZFPs) are encoded in the hundreds by the genomes of higher gence of chordates and echinoderms, and a C-terminal array of zinc vertebrates, and many act with the heterochromatin-inducing fingers (ZNF) with sequence-specific DNA-binding potential (Urru- KAP1 as repressors of transposable elements (TEs) during early tia, 2003; Birtle & Ponting, 2006; Imbeault et al, 2017). KZFP genes embryogenesis. Yet, their widespread expression in adult tissues multiplied by gene and segment duplication to count today more and enrichment at other genetic loci indicate additional roles. than 350 and 700 representatives in the human and mouse Here, we characterized the protein interactome of 101 of the ~350 genomes, respectively (Urrutia, 2003; Kauzlaric et al, 2017). A human KZFPs. Consistent with their targeting of TEs, most KZFPs majority of human KZFPs including all primate-restricted family conserved up to placental mammals essentially recruit KAP1 and members target sequences derived from TEs, that is, DNA trans- associated effectors. In contrast, a subset of more ancient KZFPs posons, ERVs (endogenous retroviruses), LINEs, SINEs (long and rather interacts with factors related to functions such as genome short interspersed nuclear elements, respectively), or SVAs (SINE- architecture or RNA processing. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -
Download Validation Data
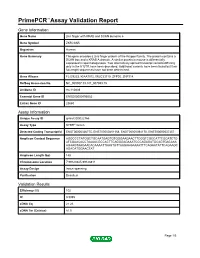
PrimePCR™Assay Validation Report Gene Information Gene Name zinc finger with KRAB and SCAN domains 5 Gene Symbol ZKSCAN5 Organism Human Gene Summary This gene encodes a zinc finger protein of the Kruppel family. The protein contains a SCAN box and a KRAB A domain. A similar protein in mouse is differentially expressed in spermatogenesis. Two alternatively spliced transcript variants differing only in the 5' UTR have been described. Additional variants have been found but their full-length sequences have not been determined. Gene Aliases FLJ39233, KIAA1015, MGC33710, ZFP95, ZNF914 RefSeq Accession No. NC_000007.13, NT_007933.15 UniGene ID Hs.110839 Ensembl Gene ID ENSG00000196652 Entrez Gene ID 23660 Assay Information Unique Assay ID qHsaCID0022766 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000326775, ENST00000451158, ENST00000394170, ENST00000537357 Amplicon Context Sequence AGGCCCTATGGCTGCAATGAGTGTGGGAAGAACTTCGGTCGCCATTCGCATCTG ATCGAACACCTAAAACGCCACTTCAGGGAGAAATCCCAGAGATGCAGTGACAAA AGAAGTAAGAACACAAAATTAAGTGTTAAGAAGAAAATTTCAGAATATTCAGAAGC AGACATGGAACTAT Amplicon Length (bp) 148 Chromosome Location 7:99123945-99128811 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 102 R2 0.9985 cDNA Cq 21.26 cDNA Tm (Celsius) 81.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 34.61 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report ZKSCAN5, Human Amplification Plot Amplification of cDNA generated from -

Mai Muudatuntuu Ti on Man Mini
MAIMUUDATUNTUU US009809854B2 TI ON MAN MINI (12 ) United States Patent ( 10 ) Patent No. : US 9 ,809 ,854 B2 Crow et al. (45 ) Date of Patent : Nov . 7 , 2017 Whitehead et al. (2005 ) Variation in tissue - specific gene expression ( 54 ) BIOMARKERS FOR DISEASE ACTIVITY among natural populations. Genome Biology, 6 :R13 . * AND CLINICAL MANIFESTATIONS Villanueva et al. ( 2011 ) Netting Neutrophils Induce Endothelial SYSTEMIC LUPUS ERYTHEMATOSUS Damage , Infiltrate Tissues, and Expose Immunostimulatory Mol ecules in Systemic Lupus Erythematosus . The Journal of Immunol @(71 ) Applicant: NEW YORK SOCIETY FOR THE ogy , 187 : 538 - 552 . * RUPTURED AND CRIPPLED Bijl et al. (2001 ) Fas expression on peripheral blood lymphocytes in MAINTAINING THE HOSPITAL , systemic lupus erythematosus ( SLE ) : relation to lymphocyte acti vation and disease activity . Lupus, 10 :866 - 872 . * New York , NY (US ) Crow et al . (2003 ) Microarray analysis of gene expression in lupus. Arthritis Research and Therapy , 5 :279 - 287 . * @(72 ) Inventors : Mary K . Crow , New York , NY (US ) ; Baechler et al . ( 2003 ) Interferon - inducible gene expression signa Mikhail Olferiev , Mount Kisco , NY ture in peripheral blood cells of patients with severe lupus . PNAS , (US ) 100 ( 5 ) : 2610 - 2615. * GeneCards database entry for IFIT3 ( obtained from < http : / /www . ( 73 ) Assignee : NEW YORK SOCIETY FOR THE genecards. org /cgi - bin / carddisp .pl ? gene = IFIT3 > on May 26 , 2016 , RUPTURED AND CRIPPLED 15 pages ) . * Navarra et al. (2011 ) Efficacy and safety of belimumab in patients MAINTAINING THE HOSPITAL with active systemic lupus erythematosus : a randomised , placebo FOR SPECIAL SURGERY , New controlled , phase 3 trial . The Lancet , 377 :721 - 731. * York , NY (US ) Abramson et al . ( 1983 ) Arthritis Rheum . -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name transmembrane protein 59 Gene Symbol Tmem59 Organism Mouse Gene Summary Description Not Available Gene Aliases 1110001M20Rik, 3110046P06Rik, AI256529, D4Ertd20e, MTDCF1, ORF18 RefSeq Accession No. NC_000070.6, NT_039264.7 UniGene ID Mm.291192 Ensembl Gene ID ENSMUSG00000028618 Entrez Gene ID 56374 Assay Information Unique Assay ID qMmuCED0047467 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000030361, ENSMUST00000128123, ENSMUST00000106753 Amplicon Context Sequence GAGACAAGAACAACTCATGTCCCTGATGCCAAGAATGCATCTCCTCTTCCCTCTG ACTCTGGTGAGGTCGTTCTGGAGTGACATGATGGACT Amplicon Length (bp) 62 Chromosome Location 4:107187701-107190613 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9997 cDNA Cq 19.5 cDNA Tm (Celsius) 84.5 gDNA Cq 25.09 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Tmem59, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. Details for each transcript can be found on the Ensembl website at www.ensembl.org. -

Eight Common Genetic Variants Associated with Serum DHEAS Levels Suggest a Key Role in Ageing Mechanisms
Eight Common Genetic Variants Associated with Serum DHEAS Levels Suggest a Key Role in Ageing Mechanisms The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Zhai, Guangju, Alexander Teumer, Lisette Stolk, John R. B. Perry, Liesbeth Vandenput, Andrea D. Coviello, Annemarie Koster, et al. 2011. Eight common genetic variants associated with serum DHEAS levels suggest a key role in ageing mechanisms. PLoS Genetics 7(4): e1002025. Published Version doi:10.1371/journal.pgen.1002025 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:5146966 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Eight Common Genetic Variants Associated with Serum DHEAS Levels Suggest a Key Role in Ageing Mechanisms Guangju Zhai1., Alexander Teumer2., Lisette Stolk3,4., John R. B. Perry5,6., Liesbeth Vandenput7., Andrea D. Coviello8., Annemarie Koster9., Jordana T. Bell1,6, Shalender Bhasin10, Joel Eriksson7, Anna Eriksson7, Florian Ernst2, Luigi Ferrucci11, Timothy M. Frayling5, Daniel Glass1, Elin Grundberg1,12, Robin Haring13,A˚ sa K. Hedman6, Albert Hofman4,14, Douglas P. Kiel15, Heyo K. Kroemer16, Yongmei Liu17, Kathryn L. Lunetta18, Marcello Maggio19, Mattias Lorentzon7, Massimo Mangino1, David Melzer5, Iva Miljkovic20, MuTHER Consortium, Alexandra Nica12,21, Brenda W. J. H. Penninx22, Ramachandran S. Vasan8,23, Fernando Rivadeneira3,4, Kerrin S. Small1,12, Nicole Soranzo1,12, Andre´ G. Uitterlinden3,4,14, Henry Vo¨ lzke24, Scott G. -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split.