Multigenic Truncation of the Semaphorin-Plexin Pathway by a Germline Chromothriptic Rearrangement Associated with Moebius Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Extracellular Matrix Phenome Across Species
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.06.980169; this version posted March 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The extracellular matrix phenome across species Cyril Statzer1 and Collin Y. Ewald1* 1 Eidgenössische Technische Hochschule Zürich, Department of Health Sciences and Technology, Institute of Translational Medicine, Schwerzenbach-Zürich CH-8603, Switzerland *Corresponding authors: [email protected] (CYE) Keywords: Phenome, genotype-to-phenotype, matrisome, extracellular matrix, collagen, data mining. Highlights • 7.6% of the human phenome originates from variations in matrisome genes • 11’671 phenotypes are linked to matrisome genes of humans, mice, zebrafish, Drosophila, and C. elegans • Expected top ECM phenotypes are developmental, morphological and structural phenotypes • Nonobvious top ECM phenotypes include immune system, stress resilience, and age-related phenotypes 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.06.980169; this version posted March 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 Abstract 2 Extracellular matrices are essential for cellular and organismal function. Recent 3 genome-wide and phenome-wide association studies started to reveal a broad 4 spectrum of phenotypes associated with genetic variants. However, the phenome or 5 spectrum of all phenotypes associated with genetic variants in extracellular matrix 6 genes is unknown. -

Protein-Coding Variants Implicate Novel Genes Related to Lipid Homeostasis
bioRxiv preprint doi: https://doi.org/10.1101/352674; this version posted June 30, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 PROTEIN-CODING VARIANTS IMPLICATE NOVEL GENES RELATED TO LIPID HOMEOSTASIS 2 CONTRIBUTING TO BODY FAT DISTRIBUTION 3 Anne E Justice¥,1,2, Tugce Karaderi¥,3,4, Heather M Highland¥,1,5, Kristin L Young¥,1, Mariaelisa Graff¥,1, 4 Yingchang Lu¥,6,7,8, Valérie Turcot9, Paul L Auer10, Rebecca S Fine11,12,13, Xiuqing Guo14, Claudia 5 Schurmann7,8, Adelheid Lempradl15, Eirini Marouli16, Anubha Mahajan3, Thomas W Winkler17, Adam E 6 Locke18,19, Carolina Medina-Gomez20,21, Tõnu Esko11,13,22, Sailaja Vedantam11,12,13, Ayush Giri23, Ken Sin 7 Lo9, Tamuno Alfred7, Poorva Mudgal24, Maggie CY Ng24,25, Nancy L Heard-Costa26,27, Mary F Feitosa28, 8 Alisa K Manning11,29,30 , Sara M Willems31, Suthesh Sivapalaratnam30,32,33, Goncalo Abecasis18, Dewan S 9 Alam34, Matthew Allison35, Philippe Amouyel36,37,38, Zorayr Arzumanyan14, Beverley Balkau39, Lisa 10 Bastarache40, Sven Bergmann41,42, Lawrence F Bielak43, Matthias Blüher44,45, Michael Boehnke18, Heiner 11 Boeing46, Eric Boerwinkle47,48, Carsten A Böger49, Jette Bork-Jensen50, Erwin P Bottinger7, Donald W 12 Bowden24,25,51, Ivan Brandslund52,53, Linda Broer21, Amber A Burt54, Adam S Butterworth55,56, Mark J 13 Caulfield16,57, Giancarlo Cesana58, John C Chambers59,60,61,62,63, Daniel I Chasman11,64,65,66, Yii-Der Ida 14 Chen14, Rajiv Chowdhury55, Cramer Christensen67, -

Different Species Common Arthritis
High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species This information is current as Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz of September 27, 2021. Ashgari and Saleh M. Ibrahim J Immunol 2009; 182:3016-3023; ; doi: 10.4049/jimmunol.0803005 http://www.jimmunol.org/content/182/5/3016 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2009/02/18/182.5.3016.DC1 Material References This article cites 36 articles, 9 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/182/5/3016.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species1 Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz Ashgari, and Saleh M. -

Functional Specialization of Human Salivary Glands and Origins of Proteins Intrinsic to Human Saliva
UCSF UC San Francisco Previously Published Works Title Functional Specialization of Human Salivary Glands and Origins of Proteins Intrinsic to Human Saliva. Permalink https://escholarship.org/uc/item/95h5g8mq Journal Cell reports, 33(7) ISSN 2211-1247 Authors Saitou, Marie Gaylord, Eliza A Xu, Erica et al. Publication Date 2020-11-01 DOI 10.1016/j.celrep.2020.108402 Peer reviewed eScholarship.org Powered by the California Digital Library University of California HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Cell Rep Manuscript Author . Author manuscript; Manuscript Author available in PMC 2020 November 30. Published in final edited form as: Cell Rep. 2020 November 17; 33(7): 108402. doi:10.1016/j.celrep.2020.108402. Functional Specialization of Human Salivary Glands and Origins of Proteins Intrinsic to Human Saliva Marie Saitou1,2,3, Eliza A. Gaylord4, Erica Xu1,7, Alison J. May4, Lubov Neznanova5, Sara Nathan4, Anissa Grawe4, Jolie Chang6, William Ryan6, Stefan Ruhl5,*, Sarah M. Knox4,*, Omer Gokcumen1,8,* 1Department of Biological Sciences, University at Buffalo, The State University of New York, Buffalo, NY, U.S.A 2Section of Genetic Medicine, Department of Medicine, University of Chicago, Chicago, IL, U.S.A 3Faculty of Biosciences, Norwegian University of Life Sciences, Ås, Viken, Norway 4Program in Craniofacial Biology, Department of Cell and Tissue Biology, School of Dentistry, University of California, San Francisco, CA, U.S.A 5Department of Oral Biology, School of Dental Medicine, University at Buffalo, The State University of New York, Buffalo, NY, U.S.A 6Department of Otolaryngology, School of Medicine, University of California, San Francisco, CA, U.S.A 7Present address: Weill-Cornell Medical College, Physiology and Biophysics Department 8Lead Contact SUMMARY Salivary proteins are essential for maintaining health in the oral cavity and proximal digestive tract, and they serve as potential diagnostic markers for monitoring human health and disease. -

Short Article Semaphorin-Plexin Signaling Guides Patterning of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Developmental Cell, Vol. 7, 117–123, July, 2004, Copyright 2004 by Cell Press Semaphorin-Plexin Signaling Short Article Guides Patterning of the Developing Vasculature Jesu´ s Torres-Va´ zquez,1,5 Aaron D. Gitler,2,5 receptors (Tessier-Lavigne and Goodman, 1996). The Sherri D. Fraser,3,5 Jason D. Berk,1 semaphorin family of ligands and their plexin receptors Van N. Pham,1 Mark C. Fishman,4,7 are key players in neuronal pathfinding. Semaphorins Sarah Childs,3,6 Jonathan A. Epstein,2,6 inhibit migration of plexin-expressing neuronal growth and Brant M. Weinstein1,6,* cones, restricting their navigation pathways (Tamag- 1Laboratory of Molecular Genetics none and Comoglio, 2000). We show here that the endo- NICHD thelial receptor PlexinD1 plays a similar role during vas- National Institutes of Health cular patterning. In the developing trunk, angiogenic Bethesda, Maryland 20892 intersegmental vessels extend near somite boundaries 2 Cardiovascular Division (Isogai et al., 2001; Childs et al., 2002). Loss of plxnD1 University of Pennsylvania function via morpholino injection or in zebrafish out of Philadelphia, Pennsylvania 19104 bounds (obd) mutants (Childs et al., 2002) causes dra- 3 Department of Biochemistry and Molecular matic mispatterning of these vessels, which are no Biology and Smooth Muscle Research Group longer restricted to growth near intersomitic boundaries. University of Calgary Somites flanking intersegmental vessels express sema- Calgary, Alberta T2N 4N1 phorins (Roos et al., 1999; Yee et al., 1999), and reducing Canada the function of these semaphorins causes similar inter- 4 Cardiovascular Research Center segmental vessel patterning defects. -

A Supplemental Fig S1| PLXND1 Genomic Locus. A, Image from the UCSC Genome Browser (Grch37/Hg19). PLXND1 Gene Is Shown in Dark B
a hg19 20 kb Scale chr3: 129,330,000 129,325,000 129,320,000 129,315,000 129,310,000 129,305,000 129,300,000 129,295,000 129,290,000 129,285,000 129,280,000 129,275,000 P1 P2 P3 P4 P+ Basic Gene Annotation Set from GENCODE Version 24lift37 (Ensembl 83) PLXND1 RN7SL752P PLXND1 10 HeLa-S3 whole cell polyA+ RNA-seq Minus signal Rep 1 from ENCODE/CSHL 1 10 HeLa-S3 whole cell polyA+ RNA-seq Minus signal Rep 2 from ENCODE/CSHL 1 10 HeLa-S3 whole cell polyA+ RNA-seq Plus signal Rep 1 from ENCODE/CSHL 1 10 HeLa-S3 whole cell polyA+ RNA-seq Plus signal Rep 2 from ENCODE/CSHL 1 10 HCT-116 200 bp paired read RNA-seq Signal Rep 1 from ENCODE/Caltech 1 10 HCT-116 200 bp paired read RNA-seq Signal Rep 2 from ENCODE/Caltech 1 Supplemental Fig S1| PLXND1 genomic locus. a, Image from the UCSC Genome Browser (GRCh37/hg19). PLXND1 gene is shown in dark blue (exons are represented as thick boxes and introns as lines). sgRNA pairs are depicted (P1, P2, P3, P4, P+) around exon 1. Whole-cell RNA expression in HeLa (ENCODE/Cold Spring Harbor Lab; plus and minus strand) and HCT116 (ENCODE/Caltech). HeLa (M3814) (n=3) Fraction of plexin D1 negative cells Standard Average deviation Vehicle 0.909 0.013 P+ 300nM 0.896 0.017 P1.1 P1.2 Vehicle 0.013 0.015 P1.1 P2.1 P2.2 300nM 0.000 0.008 Vehicle 0.009 0.005 P3.1 P3.2 P1.2 300nM 0.004 0.007 P4.1 P4.2 400 bp Vehicle 0.004 0.011 P+ P2.1 300nM 0.002 0.009 PLXND1 gene Vehicle 0.006 0.009 chr3:129,325,797 TSS Exon 1 Intron 1 Exon 2 P2.2 300nM 0.003 0.009 Vehicle 0.006 0.012 P3.1 300nM 0.002 0.007 Vehicle 0.001 0.009 P3.2 300nM 0.000 0.008 Vehicle 0.016 0.015 P4.1 300nM 0.012 0.012 Vehicle 0.016 0.014 P4.2 300nM 0.010 0.008 Supplemental Fig S2| Effect of single sgRNAs around PLXND1 first exon. -

GIPC Proteins Negatively Modulate Plexind1 Signaling During Vascular Development
RESEARCH ARTICLE GIPC proteins negatively modulate Plexind1 signaling during vascular development Jorge Carretero-Ortega1*, Zinal Chhangawala1, Shane Hunt1, Carlos Narvaez1, Javier Mene´ ndez-Gonza´ lez1, Carl M Gay1, Tomasz Zygmunt1, Xiaochun Li2, Jesu´s Torres-Va´ zquez1* 1Department of Cell Biology, Skirball Institute of Biomolecular Medicine, New York University Langone Medical Center, New York, United States; 2Department of Population Health, New York University School of Medicine, New York, United States Abstract Semaphorins (SEMAs) and their Plexin (PLXN) receptors are central regulators of metazoan cellular communication. SEMA-PLXND1 signaling plays important roles in cardiovascular, nervous, and immune system development, and cancer biology. However, little is known about the molecular mechanisms that modulate SEMA-PLXND1 signaling. As PLXND1 associates with GIPC family endocytic adaptors, we evaluated the requirement for the molecular determinants of their association and PLXND1’s vascular role. Zebrafish that endogenously express a Plxnd1 receptor with a predicted impairment in GIPC binding exhibit low penetrance angiogenesis deficits and antiangiogenic drug hypersensitivity. Moreover, gipc mutant fish show angiogenic impairments that are ameliorated by reducing Plxnd1 signaling. Finally, GIPC depletion potentiates SEMA-PLXND1 signaling in cultured endothelial cells. These findings expand the vascular roles of GIPCs beyond those of the Vascular Endothelial Growth Factor (VEGF)-dependent, proangiogenic GIPC1- Neuropilin 1 complex, recasting GIPCs as negative modulators of antiangiogenic PLXND1 signaling *For correspondence: and suggest that PLXND1 trafficking shapes vascular development. [email protected] (JC- DOI: https://doi.org/10.7554/eLife.30454.001 O); [email protected] (JT-V) Competing interests: The authors declare that no Introduction competing interests exist. Angiogenic sprouting, the formation of new vessels via branching of pre-existing ones, drives most Funding: See page 29 of the life-sustaining expansion of the vascular tree. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands That Promote Axonal Growth
Research Article: New Research Development Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands that Promote Axonal Growth Jeremy S. Toma,1 Konstantina Karamboulas,1,ª Matthew J. Carr,1,2,ª Adelaida Kolaj,1,3 Scott A. Yuzwa,1 Neemat Mahmud,1,3 Mekayla A. Storer,1 David R. Kaplan,1,2,4 and Freda D. Miller1,2,3,4 https://doi.org/10.1523/ENEURO.0066-20.2020 1Program in Neurosciences and Mental Health, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada, 2Institute of Medical Sciences University of Toronto, Toronto, Ontario M5G 1A8, Canada, 3Department of Physiology, University of Toronto, Toronto, Ontario M5G 1A8, Canada, and 4Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5G 1A8, Canada Abstract Peripheral nerves provide a supportive growth environment for developing and regenerating axons and are es- sential for maintenance and repair of many non-neural tissues. This capacity has largely been ascribed to paracrine factors secreted by nerve-resident Schwann cells. Here, we used single-cell transcriptional profiling to identify ligands made by different injured rodent nerve cell types and have combined this with cell-surface mass spectrometry to computationally model potential paracrine interactions with peripheral neurons. These analyses show that peripheral nerves make many ligands predicted to act on peripheral and CNS neurons, in- cluding known and previously uncharacterized ligands. While Schwann cells are an important ligand source within injured nerves, more than half of the predicted ligands are made by nerve-resident mesenchymal cells, including the endoneurial cells most closely associated with peripheral axons. At least three of these mesen- chymal ligands, ANGPT1, CCL11, and VEGFC, promote growth when locally applied on sympathetic axons. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptNature. Author ManuscriptAuthor manuscript; Author Manuscript available in PMC 2015 November 28. Published in final edited form as: Nature. 2015 May 28; 521(7553): 520–524. doi:10.1038/nature14269. Global genetic analysis in mice unveils central role for cilia in congenital heart disease You Li1,8, Nikolai T. Klena1,8, George C Gabriel1,8, Xiaoqin Liu1,7, Andrew J. Kim1, Kristi Lemke1, Yu Chen1, Bishwanath Chatterjee1, William Devine2, Rama Rao Damerla1, Chien- fu Chang1, Hisato Yagi1, Jovenal T. San Agustin5, Mohamed Thahir3,4, Shane Anderton1, Caroline Lawhead1, Anita Vescovi1, Herbert Pratt5, Judy Morgan6, Leslie Haynes6, Cynthia L. Smith6, Janan T. Eppig6, Laura Reinholdt6, Richard Francis1, Linda Leatherbury7, Madhavi K. Ganapathiraju3,4, Kimimasa Tobita1, Gregory J. Pazour5, and Cecilia W. Lo1,9 1Department of Developmental Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 2Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA 3Department of Biomedical Informatics, University of Pittsburgh School of Medicine, Pittsburgh, PA 4Intelligent Systems Program, School of Arts and Sciences, University of Pittsburgh, Pittsburgh, PA 9Corresponding author. [email protected] Phone: 412-692-9901, Mailing address: Dept of Developmental Biology, Rangos Research Center, 530 45th St, Pittsburgh, PA, 15201. 8Co-first authors Author Contributions: Study design: CWL ENU mutagenesis, line cryopreservation, JAX strain datasheet construction: -
PDF-Document
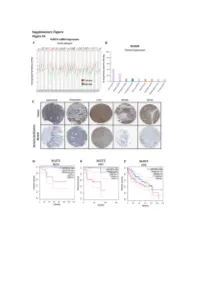
Figure S1: NUDT5 is indicative of poor prognosis and aggressive cancer growth. (A) NUDT5 mRNA expression across full TGCA database, data for tumour (red) and normal (green) tissue from the same tissue origin are shown using TGCA patient datasets with GEPIA interactive webserver [61]. Datasets where NUDT5 mRNA levels are elevated in tumour versus normal as highlighted in red, significantly higher in normal versus tumour are marked in green. Abbreviations of the tumour datasets are given in Table S1. (B) The percentage of positive staining for NUDT5 protein expression in cancer datasets (Human Protein Atlas database [62]). (C) Representative histological staining of NUDT5 in tumour versus normal patient samples from colorectal, pancreatic, liver, breast and renal samples (Human Protein Atlas database [62]). Metadata regarding each of the images are given in Table S2. Kaplan Meyer survival curves stratifying patients based on NUDT5 mRNA expression in (D) Kidney chromophobe (KICH). (E) Adrenalcorticocal carcinoma (ACC). (F) Liver hepatocellular carcinoma (LIHG) (G) Brain lower grade carcinoma (LGG) (H) Kidney renal clear cell carcinoma (KIRC) (I) Kidney renal papillary cell carcinoma (KIRP) were carried out using TGCA database with GEPIA interactive webserver [61]. (J) Recurrence and metastasis analysis in NUDT5 mRNA high expressing patient cohorts from BRCA dataset shown in Figure S1A. (K) Full western blot of NUDT5, PARP1 and FLAG protein levels in NUDT5RES and NUDT5KD cell clones. Figure 2. Analysis of the gene expression changes in 3D compared to 2D culture conditions. (A) PCA analysis of all RNA samples (Table S3) from T47D cells cultured in 2D and 3D conditions. (B) PCA analysis of RNA samples from T47D cells highlighted based on culture conditions. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein