Supplementary Materials for “Mash: Fast Genome and Metagenome Distance Estimation Using Minhash”
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SUSANNE BRUNKHORST, D V M (B)(6), (B)(7)(C)
SBRUNKHORST United States Department of Agriculture Animal and Plant Health Inspection Service 2016082567931581 Insp_id Inspection Report University Of Tennesee - Chattanooga Customer ID: 29 975 East Third St. Certificate: 63-R-0001 Box 339 Site: 001 College Of Medicine COLLEGE OF MEDICINE Chattanooga, TN 37403 Type: ROUTINE INSPECTION Date: 20-SEP-2016 No non-compliant items identified during this inspection. This inspection and exit interview were conducted with undersigned facility representative. SUSANNE BRUNKHORST, D V M Prepared By: Date: SUSANNE BRUNKHORST USDA, APHIS, Animal Care 20-SEP-2016 Title: VETERINARY MEDICAL OFFICER 1076 Received By: (b)(6), (b)(7)(c) Date: Title: FACILITY REPRESENTATIVE 20-SEP-2016 Page 1 of 1 United States Department of Agriculture Customer: 29 Animal and Plant Health Inspection Service Inspection Date: 20-SEP-16 Animal Inspected at Last Inspection Cust No Cert No Site Site Name Inspection 29 63-R-0001 001 UNIVERSITY OF TENNESEE - 20-SEP-16 CHATTANOOGA Count Species 000000 None 000000 Total United States Department of Agriculture Customer: 29 Animal and Plant Health Inspection Service Inspection Date: 24-AUG-15 Animal Inspected at Last Inspection Cust No Cert No Site Site Name Inspection 29 63-R-0001 001 UNIVERSITY OF TENNESEE - 24-AUG-15 CHATTANOOGA Count Species 000000 None 000000 Total United States Department of Agriculture Customer: 851 Animal and Plant Health Inspection Service Inspection Date: 04-FEB-16 Animal Inspected at Last Inspection Cust No Cert No Site Site Name Inspection 851 63-R-0002 -

The Taxonomy of Primates in the Laboratory Context
P0800261_01 7/14/05 8:00 AM Page 3 C HAPTER 1 The Taxonomy of Primates T HE T in the Laboratory Context AXONOMY OF P Colin Groves RIMATES School of Archaeology and Anthropology, Australian National University, Canberra, ACT 0200, Australia 3 What are species? D Taxonomy: EFINITION OF THE The biological Organizing nature species concept Taxonomy means classifying organisms. It is nowadays commonly used as a synonym for systematics, though Disagreement as to what precisely constitutes a species P strictly speaking systematics is a much broader sphere is to be expected, given that the concept serves so many RIMATE of interest – interrelationships, and biodiversity. At the functions (Vane-Wright, 1992). We may be interested basis of taxonomy lies that much-debated concept, the in classification as such, or in the evolutionary implica- species. tions of species; in the theory of species, or in simply M ODEL Because there is so much misunderstanding about how to recognize them; or in their reproductive, phys- what a species is, it is necessary to give some space to iological, or husbandry status. discussion of the concept. The importance of what we Most non-specialists probably have some vague mean by the word “species” goes way beyond taxonomy idea that species are defined by not interbreeding with as such: it affects such diverse fields as genetics, biogeog- each other; usually, that hybrids between different species raphy, population biology, ecology, ethology, and bio- are sterile, or that they are incapable of hybridizing at diversity; in an era in which threats to the natural all. Such an impression ultimately derives from the def- world and its biodiversity are accelerating, it affects inition by Mayr (1940), whereby species are “groups of conservation strategies (Rojas, 1992). -

NO2N Import Into Containment Any New Organism That Is Not Genetically Modified
NO2N Import into containment any new organism that is not genetically modified Application title: Importation of specified “new” mammal species into containment at Wellington Zoo, and other zoos, to aid conservation though sustainable display, captive breeding and / or the conservation of genetic material Applicant organisation: Wellington Zoo Trust, 200 Daniell Street, Newtown, Wellington Please provide a brief summary of the purpose of the application (255 characters or less, including spaces) To import into containment 28 mammal species for captive breeding, display, educational presentations and to contribute to conservation by exposing visitors to conservation issues and the conservation of genetic material through breeding PLEASE CONTACT ERMA NEW ZEALAND BEFORE SUBMITTING YOUR APPLICATION Please clearly identify any confidential information and attach as a separate appendix. Please check and complete the following before submitting your application: All sections completed Yes Appendices enclosed NA Confidential information identified and enclosed separately NA Copies of references attached Yes Application signed and dated Yes Electronic copy of application e-mailed to Yes ERMA New Zealand Signed: Date: 20 Customhouse Quay Cnr Waring Taylor and Customhouse Quay PO Box 131, Wellington Phone: 04 916 2426 Fax: 04 914 0433 Email: [email protected] Website: www.ermanz.govt.nz NO2N: Application to import into containment any new organism that is not genetically modified Section One – Applicant details Name and details of the organisation -

Evolutionary Stasis of the Pseudoautosomal Boundary In
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates Rylan Shearn, Alison E Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaître, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel Ab Marais To cite this version: Rylan Shearn, Alison E Wright, Sylvain Mousset, Corinne Régis, Simon Penel, et al.. Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates. eLife, eLife Sciences Publication, 2020, 9, 10.7554/eLife.63650. hal-03064964 HAL Id: hal-03064964 https://hal.archives-ouvertes.fr/hal-03064964 Submitted on 14 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. SHORT REPORT Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates Rylan Shearn1, Alison E Wright2, Sylvain Mousset1,3, Corinne Re´ gis1, Simon Penel1, Jean-Franc¸ois Lemaitre1, Guillaume Douay4, Brigitte Crouau-Roy5, Emilie Lecompte5, Gabriel AB Marais1,6* 1Laboratoire Biome´trie et Biologie Evolutive, CNRS / Univ. Lyon 1, Villeurbanne, France; 2Department of Animal and Plant Sciences, University of Sheffield, Sheffield, United Kingdom; 3Faculty of Mathematics, University of Vienna, Vienna, Austria; 4Zoo de Lyon, Lyon, France; 5Laboratoire Evolution et Diversite´ Biologique, CNRS / Univ. Toulouse, Toulouse, France; 6LEAF-Linking Landscape, Environment, Agriculture and Food Dept, Instituto Superior de Agronomia, Universidade de Lisboa, Lisbon, Portugal Abstract Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. -

Supplementary Materials For
Supplementary Materials for Contrasted sex chromosome evolution in primates with and without sexual dimorphism Rylan Shearn, Emilie Lecompte, Corinne Régis, Sylvain Mousset, Simon Penel, Guillaume Douay, Brigitte Crouau-Roy, Gabriel A.B. Marais Correspondence to: [email protected] This PDF file includes: Supplementary Text S1 to S2 Figs. S1 to S2 Tables S1 1 Supplementary Text Text S1: Regions of the strepsirrhine X chromosomes with unusual male:female coverage ratio In Fig. 1, both lemur X chromosomes exhibit regions with male:female coverage ratio close to 1 (shown in grey) in their X-specific parts, where a ratio of 0.5 is expected. The gray mouse lemur has five such regions, the northern greater galago three. The dot plots of the lemur and the human X chromosomes (see Fig. 1 and S1) clearly show that little or no homologous genes are found in those regions, which suggest that they may be homologous to other human chromosomes. This would be consistent with the male:female coverage ratio of 1, typical of autosmal regions, that we found for these regions. To explore this possibility, we extracted the sequences of those regions and performed a tblastn against all the human proteins (human genome version GRCh38). In case of isoforms, the longest protein was kept so that a human gene was present only once. We then filtered the tblastn results by keeping only hits with >80% similarity (based on average nucleotide divergence between lemurs and humans) and e-value < 10-9. From those, we kept human proteins covered by hits to >80% using SiLix (Miele, Penel, & Duret, 2011). -

Animal Inspected at Last Inspection
United States Department of Agriculture Customer: 3432 Animal and Plant Health Inspection Service Inspection Date: 10-AUG-16 Animal Inspected at Last Inspection Cust No Cert No Site Site Name Inspection 3432 86-C-0001 001 ARIZONA CENTER FOR NATURE 10-AUG-16 CONSERVATION Count Species 000003 Cheetah 000005 Cattle/cow/ox/watusi 000003 Mandrill *Male 000006 Hamadryas baboon 000004 Grevys zebra 000008 Thomsons gazelle 000002 Cape Porcupine 000002 Lion 000002 African hunting dog 000002 Tiger 000008 Common eland 000002 Spotted hyena 000001 White rhinoceros 000007 Spekes gazelle 000005 Giraffe 000004 Kirks dik-dik 000002 Fennec fox 000003 Ring-tailed lemur 000069 Total ARHYNER United States Department of Agriculture Animal and Plant Health Inspection Service 2016082567967934 Insp_id Inspection Report Arizona Center For Nature Conservation Customer ID: 3432 455 N. Galvin Parkway Certificate: 86-C-0001 Phoenix, AZ 85008 Site: 001 ARIZONA CENTER FOR NATURE CONSERVATION Type: ROUTINE INSPECTION Date: 19-OCT-2016 No non-compliant items identified during this inspection. This inspection and exit interview were conducted with the primate manager. Additional Inspectors Gwendalyn Maginnis, Veterinary Medical Officer AARON RHYNER, D V M Prepared By: Date: AARON RHYNER USDA, APHIS, Animal Care 19-OCT-2016 Title: VETERINARY MEDICAL OFFICER 6077 Received By: (b)(6), (b)(7)(c) Date: Title: FACILITY REPRESENTATIVE 19-OCT-2016 Page 1 of 1 United States Department of Agriculture Customer: 3432 Animal and Plant Health Inspection Service Inspection Date: 19-OCT-16 -
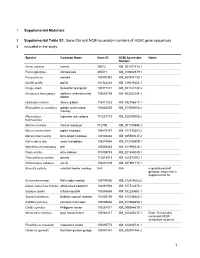
1 Supplemental Materials 1 Supplemental Table S1. Gene Ids and NCBI Accession Numbers of ACE2 Gene Sequences 2 Included In
1 Supplemental Materials 2 Supplemental Table S1. Gene IDs and NCBI accession numbers of ACE2 gene sequences 3 included in the study. Species Common Name Gene ID NCBI Accession Notes Number Homo sapiens human 59272 NM_001371415.1 Pan troglodytes chimpanzee 465511 XM_016942979.1 Pan paniscus bonobo 100970340 XM_008974180.1 Gorilla gorilla gorilla 101142534 XM_019019204.1 Pongo abelii Sumatran orangutan 100171441 NM_001131132.2 Nomascus leucogenys northern white-cheeked 100602708 XM_003261084.3 gibbon Hylobates moloch silvery gibbon 116811532 XM_032756617.1 Rhinopithecus roxellana golden snub-nosed 104664530 XM_010366065.2 monkey Piliocolobus Ugandan red colobus 111531712 XM_023199053.2 tephrosceles Macaca mulatta rhesus macaque 712790 NM_001135696.1 Macaca nemestrina pigtail macaque 105478157 XM_011735203.2 Macaca fascicularis long-tailed macaque 102130864 XM_005593037.2 Cercocebus atys sooty mangabey 105574684 XM_012035809.1 Mandrillus leucophaeus drill 105550583 XM_011995533.1 Papio anubis olive baboon 101008749 XM_021933040.1 Theropithecus gelada gelada 112615413 XM_025372062.1 Chlorocebus sabaeus vervet 103231639 XM_007991113.1 Alouatta palliata mantled howler monkey N/A N/A unpublished draft genome, sequence in supplemental file Aotus nancymaae Ma’s night monkey 105705080 XM_012434682.2 Cebus capucinus imitator white-faced capuchin 108291904 XM_017512376.1 Sapajus apella tufted capuchin 116556688 XM_032285963.1 Saimiri boliviensis Bolivian squirrel monkey 101045190 XM_010336623.1 Callithrix jacchus common marmoset 100408882 XM_017968359.1 -

Evolutionary Stasis of the Pseudoautosomal Boundary in Strepsirrhine Primates
This is a repository copy of Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/168368/ Version: Published Version Article: Shearn, R., Wright, A.E., Mousset, S. et al. (7 more authors) (2020) Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates. eLife, 9. e63650. ISSN 2050- 084X https://doi.org/10.7554/elife.63650 Reuse This article is distributed under the terms of the Creative Commons Attribution (CC BY) licence. This licence allows you to distribute, remix, tweak, and build upon the work, even commercially, as long as you credit the authors for the original work. More information and the full terms of the licence here: https://creativecommons.org/licenses/ Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ SHORT REPORT Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates Rylan Shearn1, Alison E Wright2, Sylvain Mousset1,3, Corinne Re´ gis1, Simon Penel1, Jean-Franc¸ois Lemaitre1, Guillaume Douay4, Brigitte Crouau-Roy5, Emilie Lecompte5, Gabriel AB Marais1,6* 1Laboratoire Biome´trie et Biologie Evolutive, CNRS / Univ. Lyon 1, Villeurbanne, France; 2Department of Animal and Plant Sciences, University of Sheffield, Sheffield, United Kingdom; 3Faculty of Mathematics, University of Vienna, Vienna, Austria; 4Zoo de Lyon, Lyon, France; 5Laboratoire Evolution et Diversite´ Biologique, CNRS / Univ. Toulouse, Toulouse, France; 6LEAF-Linking Landscape, Environment, Agriculture and Food Dept, Instituto Superior de Agronomia, Universidade de Lisboa, Lisbon, Portugal Abstract Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. -

BMC Evolutionary Biology Biomed Central
BMC Evolutionary Biology BioMed Central Research article Open Access Evolution of a behavior-linked microsatellite-containing element in the 5' flanking region of the primate AVPR1A gene Zoe R Donaldson*1,2,3, Fyodor A Kondrashov4, Andrea Putnam4, Yaohui Bai3, Tara L Stoinski5, Elizabeth AD Hammock6 and Larry J Young1,2,3,7 Address: 1Neuroscience Program, Emory University, Atlanta, USA, 2Center for Behavioral Neuroscience, Emory University, Atlanta, USA, 3Yerkes National Primate Research Center, Emory University, Atlanta, USA, 4Division of Biological Sciences, University of California, San Diego, La Jolla, USA, 5Zoo Atlanta, Atlanta, USA, 6Vanderbilt Kennedy Center for Research on Human Development, Nashville, USA and 7Department of Psychiatry and Behavioral Sciences, Emory University, Atlanta, USA Email: Zoe R Donaldson* - [email protected]; Fyodor A Kondrashov - [email protected]; Andrea Putnam - [email protected]; Yaohui Bai - [email protected]; Tara L Stoinski - [email protected]; Elizabeth AD Hammock - [email protected]; Larry J Young - [email protected] * Corresponding author Published: 23 June 2008 Received: 6 March 2008 Accepted: 23 June 2008 BMC Evolutionary Biology 2008, 8:180 doi:10.1186/1471-2148-8-180 This article is available from: http://www.biomedcentral.com/1471-2148/8/180 © 2008 Donaldson et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The arginine vasopressin V1a receptor (V1aR) modulates social cognition and behavior in a wide variety of species. -

JAALAS 2020 Board Examinations Past Or Present Information Presented Is Believed to Be Reliable and Correct Though Not Guaranteed for Accuracy Francis J
DISCLAIMERS ACLAM has not reviewed or sanctioned this presentation No information presented is known to be included in the ACLAM JAALAS 2020 Board Examinations past or present Information presented is believed to be reliable and correct though not guaranteed for accuracy Francis J. Sun, DVM, DACLAM, MBA Not every article is included in this review Laboratory Animal Workshop 2021 I used the 2nd edition Blue Book 1 2 General study tips ACLAM certification site Know your Role Delineation Document Laboratory Animal Medicine, 3rd Edition Allocate your time accordingly The 3rd edition of Laboratory Animal Medicine is currently available in electronic format, with the hard cover version available soon. As it is an Trust yourself with your study habits ACLAM sanctioned textbook, it is considered fair game for the 2016 exam. When studying, please realize that there are over 3,000 questions Groups vs individual in our database. It will take the exam committee longer than a single Practice exams or note cards exam cycle to update every reference; however all questions for the 2016 exam that have the 2nd edition of the Blue Book as a reference Rest and health lifestyle are more beneficial for the last couple of will be fact checked against the 3rd edition of the Blue Book and days than cramming updated as necessary. Exam Material Cut Off Date The annual ACLAM Exam may contain questions on materials from any relevant publications up to Dec 31 of the year prior to the exam (Dec 31, 2019 for the 2020 exam). 3 4 ACLAM certification site ACLAM certification site ACLAM 2021 Certification Examination Reference List : This reference list provides the sources of the majority of the examination Domain 1: Management of Spontaneous and Experimentally questions for the 2021 exam. -

<I>Otolemur Garnettii</I>
The University of Southern Mississippi The Aquila Digital Community Honors Theses Honors College Spring 5-2017 Predatory Nature and Food Preferences Among Captive Otolemur garnettii Morgan N. Ruby University of Southern Mississippi Follow this and additional works at: https://aquila.usm.edu/honors_theses Part of the Animal Studies Commons Recommended Citation Ruby, Morgan N., "Predatory Nature and Food Preferences Among Captive Otolemur garnettii" (2017). Honors Theses. 493. https://aquila.usm.edu/honors_theses/493 This Honors College Thesis is brought to you for free and open access by the Honors College at The Aquila Digital Community. It has been accepted for inclusion in Honors Theses by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. The University of Southern Mississippi Predatory Nature and Food Preferences among Captive Otolemur garnettii by Morgan Ruby A Thesis Submitted to the Honors College of The University of Southern Mississippi in Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in the Department of Biological Sciences May 2017 ii Approved by ________________________________ Dr. B. Katherine Smith Department of Anthropology ________________________________ Dr. Ann Marie Kinnell Department of Anthropology and Sociology, Chair ________________________________ Dr. Ellen Weinauer Honors College, Dean iii Abstract Observing the predatory nature in primates has yielded knowledge pertaining to their biology and evolutionary pathways; however, not many studies have focused on the complexities of their food preferences. This thesis focuses on food preferences among Garnett’s Greater Bushbaby (Otolemur garnettii): a small-bodied nocturnal primate native to Central and Southern Africa. Presented food options were raisins: dried mealworms and raisins: live mealworms. -

Food Choice and Nutritional Analysis of Frugivorous and Insectivorous Diets in Captive Northern Greater Galagos (Otolemur Garnettii)
Food choice and nutritional analysis of frugivorous and insectivorous diets in captive Northern greater galagos (Otolemur garnettii) 1 2 2 3 1 4 5 5 Mead A. Krowka , B. Katherine Smith , Sydnie Bianchi , James E. Loudon , Michaela E. Howells , Andres M. Gomez , Oliver C.C. Paine , Matt Sponheimer 11DepartmentDepartment ofof Anthropology,Anthropology, UniversityUniversity ofof NorthNorth CarolinaCarolina--Wilmington,Wilmington, 22SchoolDepartment of Social of Anthropology Science and Globaland Sociology, Studies, TheThe UniversityUniversity ofof SouthernSouthern Mississippi,Mississippi, 33DepartmentDepartment ofof Anthropology,Anthropology, EastEast Carolina University, 4Department of Animal Science, University of Minnesota, 5Department of Anthropology, University of Colorado, Boulder Objectives Insectivorous Diet Hypotheses • The galagos consumed greater quantities of redworms and nightcrawlers • Assess the dietary preferences of a captive colony of (P<0.0001) compared to crickets and mealworms (Figure 5) H : The galagos will consume higher proportions of fruits high in non- Northern greater galagos (Otolemur garnettii) 1 • Redworms and nightcrawlers were higher in NSC (P<0.01) and lower in structural carbohydrates NDF (P<0.01) compared to crickets and mealworms (Table 1) • Understand the nutritional properties that impact O. H : The galagos will consume higher proportions fruits and invertebrates • Mealworms were higher in CF (P<0.0001) compared to all other garnettii food choice on an experimental “frugivorous” 2 with low levels of dietary inhibitors (i.e. acid detergent fiber and invertebrates (Table 1; Figure 6) and “insectivorous” diet neutral detergent fiber) • Crickets and nightcrawlers were higher in CP (P<0.0001) compared to the Introduction remaining invertebrates (Table 1) Figure 2: Examples of food items (left to right: raspberries, tamarind, crickets) Figure 5.