Quantum Chemical Study of the Thermochemical Properties of Organophosphorous Compounds A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Monocrotophos Proposed AEGL Document
MONOCROTOPHOS Page 1 of 32 Proposed 10/2009 v.1 1 2 3 4 ACUTE EXPOSURE GUIDELINE LEVELS 5 (AEGLs) 6 7 8 PROPOSED 9 10 11 12 13 MONOCROTOPHOS 14 (CAS Reg. No. 6923-22-4) 15 16 17 18 19 20 MONOCROTOPHOS Page 2 of 32 Proposed 10/2009 v.1 1 2 PREFACE 3 4 Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 5 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous 6 Substances (NAC/AEGL Committee) has been established to identify, review and interpret 7 relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic 8 chemicals. 9 10 AEGLs represent threshold exposure limits for the general public and are applicable to 11 emergency exposure periods ranging from 10 minutes to 8 hours. Three levels C AEGL-1, 12 AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 13 hour, 4 hours, and 8 hours) and are distinguished by varying degree of severity of toxic effects. 14 The three AEGLs are defined as follows: 15 16 AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per 17 cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general 18 population, including susceptible individuals, could experience notable discomfort, irritation, or 19 certain asymptomatic, non-sensory effects. However, the effects are not disabling and are 20 transient and reversible upon cessation of exposure. -

Downloads/Pgm Hydrus1d/HYDRUS-4.16.Pdf (Accessed on 20 April 2019)
water Article Assessment of the Environmental Risk of Pesticides Leaching at the Watershed Scale under Arid Climatic Conditions and Low Recharge Rates Hesham M. Ibrahim 1,2,* and Ali M. Al-Turki 1 1 Department of Soil Science, College of Food and Agricultural Sciences, King Saud University, P.O. Box 2460, Riyadh 11451, Saudi Arabia; [email protected] 2 Department of Soils and Water, Faculty of Agriculture, Suez Canal University, Ismailia 41522, Egypt * Correspondence: [email protected]; Tel.: +966-501728656 Received: 5 January 2020; Accepted: 2 February 2020; Published: 5 February 2020 Abstract: The assessment of the vulnerability of soil and groundwater resources to pesticide contamination is important to reduce the risk of environmental pollution. The applicability of the expanded attenuation factor (EAF) to assess leaching potential of 30 pesticides was investigated under 1 four recharge rates (0.0003–0.002 m d− ) in the arid environment of the Jazan watershed. EAF results revealed that Picloram, Carbofuran, Monocrotophos, and 2,4-D pesticides showed high leaching potential, mainly because of their low KOC, and relatively longer t1/2. In addition, medium leaching potential was observed with six more pesticides (Atrazine, Aldicarb, Simazine, Methomyl, Oxamyl, and Lindane). Regardless of the recharge rate, all other pesticides showed a very low leaching potential in the Jazan watershed. Sensitivity analysis revealed that the output of the EAF index is most sensitive to the fraction of organic carbon ( foc), water content at field capacity (θFC ), recharge rate (q), and partition coefficient (KOC), and least sensitive to soil bulk density (ρb) and air-filled porosity (na). -

Neuroactive Insecticides: Targets, Selectivity, Resistance, and Secondary Effects
EN58CH06-Casida ARI 5 December 2012 8:11 Neuroactive Insecticides: Targets, Selectivity, Resistance, and Secondary Effects John E. Casida1,∗ and Kathleen A. Durkin2 1Environmental Chemistry and Toxicology Laboratory, Department of Environmental Science, Policy, and Management, 2Molecular Graphics and Computational Facility, College of Chemistry, University of California, Berkeley, California 94720; email: [email protected], [email protected] Annu. Rev. Entomol. 2013. 58:99–117 Keywords The Annual Review of Entomology is online at acetylcholinesterase, calcium channels, GABAA receptor, nicotinic ento.annualreviews.org receptor, secondary targets, sodium channel This article’s doi: 10.1146/annurev-ento-120811-153645 Abstract Copyright c 2013 by Annual Reviews. Neuroactive insecticides are the principal means of protecting crops, people, All rights reserved livestock, and pets from pest insect attack and disease transmission. Cur- ∗ Corresponding author rently, the four major nerve targets are acetylcholinesterase for organophos- phates and methylcarbamates, the nicotinic acetylcholine receptor for neonicotinoids, the γ-aminobutyric acid receptor/chloride channel for by Public Health Information Access Project on 04/29/14. For personal use only. Annu. Rev. Entomol. 2013.58:99-117. Downloaded from www.annualreviews.org polychlorocyclohexanes and fiproles, and the voltage-gated sodium channel for pyrethroids and dichlorodiphenyltrichloroethane. Species selectivity and acquired resistance are attributable in part to structural differences in binding subsites, receptor subunit interfaces, or transmembrane regions. Additional targets are sites in the sodium channel (indoxacarb and metaflumizone), the glutamate-gated chloride channel (avermectins), the octopamine receptor (amitraz metabolite), and the calcium-activated calcium channel (diamides). Secondary toxic effects in mammals from off-target serine hydrolase inhibi- tion include organophosphate-induced delayed neuropathy and disruption of the cannabinoid system. -

Agents for Defense Against Chemical Warfare: Reactivators of Acetylcholinesterase Inhibited with Neurotoxic Organophosphorus Compounds **
Mil. Med. Sci. Lett. (Voj. Zdrav. Listy) 2015, vol. 84(3), p. 115-127 ISSN 0372-7025 DOI: 10.31482/mmsl.2015.013 REVIEW ARTICLE AGENTS FOR DEFENSE AGAINST CHEMICAL WARFARE: REACTIVATORS OF ACETYLCHOLINESTERASE INHIBITED WITH NEUROTOXIC ORGANOPHOSPHORUS COMPOUNDS ** Petronilho, E. C., Figueroa-Villar, J. D. Chemistry Engineering Section, Medicinal Chemistry Group, Military Institute of Engineering, Praça General Tibúrcio, 80, Urca, 22290-270, Rio de Janeiro, RJ, Brazil. Received 30 th April 2015. Revised 7 th July 2015. Published 4 th September 2015. Summary The chemical warfare agents and neurotoxic agents are an important threat to people all over the world, and require special attention because they are highly dangerous. Most of these agents are neurotoxic organophosphorus compounds (OP), which inhibit the enzyme acetylcholinesterase (AChE), which is responsible for controlling the transmission of nerve impulses. To be inhibited by these compounds, AChE can sometimes be reactivated using cationic oximes, which are the most used substances for this reactivation. Until today there have not been discovered agents for complete treatment of poisoning by all OPs. For this reason, the treatment of intoxicated people requires the determination of the absorbed OP, in order to select the appropriate activator, a process that usually requires long time and may cause death. Therefore, this study aims to do a review on the OPs used as chemical warfare agents and the process of inhibition and reactivation of AChE, especially to motivate the development of new agents for defense against chemical weapons, a process that is very important for protecting all humanity. Key words: acetylcholinesterase; AChE reactivators; organophosphorus; oximes; warfare agents INTRODUCTION pounds (OP), which are highly toxic, allowing their use with small quantities in order to cause seizures The use of chemical warfare agents is a major and death. -

EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water with Direct Aqueous Injection LC/MS/MS
EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water with Direct Aqueous Injection LC/MS/MS E. Michael Thurman and Imma Ferrer Center for Environmental Mass Spectrometry University of Colorado Boulder, CO, USA Confidentiality Label 1 March 20, 2012 Abstract EPA Method 538 is a new method from EPA for organophosphate pesticides in drinking water. It uses direct aqueous injection; thus, no sample preparation is needed. We use both UHPLC (Agilent 1290) and MS/MS (Agilent 6460) analysis for rapid analysis and sensitive detection with ng/L limits of detection. A second MRM is added for more reliable identification. Confidentiality Label 2 March 20, 2012 Hypothesis Direct injection of organophosphate pesticides (EPA Method 538) will work by UHPLC (Agilent Model 1290) and LC/MS/MS with Jetstream (Agilent Model 6460) with trace level detection at ng/L concentrations. Confidentiality Label 3 March 20, 2012 1. Introduction-Summary 1.1 EPA Method 538 (published in November 2009 by Shoemaker) deals with Organophosphate pesticides in drinking water (1) and one other contaminant, quinoline. 1.2 The method consists of 10 compounds: acephate, aldicarb, aldicarb sulfoxide, dicrotophos, diisopropylmethylphosphonate (DIMP), fenamiphos sulfone, fenamiphos sulfoxide, methamidophos, oxydemeton methyl, quinoline, and thiofanox with 5 labeled internal standards. 1.3 Direct aqueous injection is used with a large volume sample of 100 microliters; thus, no sample preparation is needed. 1.4 Because solid phase extraction (i.e. concentration of the sample is not carried out) suppression is mimimized in the analysis. 1.5 Part-per-Trillion Detection Limits. Confidentiality Label 4 March 20, 2012 Introduction 1.1: EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Direct Aqueous Injection by Jody Shoemaker, EPA Cincinnati, OH [email protected] 513-569-7298 Confidentiality Label 5 March 20, 2012 Introduction: 1.2. -

Acute Exposure to a Sublethal Dose of Imidacloprid and Coumaphos Enhances Olfactory Learning and Memory in the Honeybee Apis Mellifera
Invert Neurosci DOI 10.1007/s10158-012-0144-7 ORIGINAL PAPER Acute exposure to a sublethal dose of imidacloprid and coumaphos enhances olfactory learning and memory in the honeybee Apis mellifera Sally M. Williamson • Daniel D. Baker • Geraldine A. Wright Received: 8 October 2012 / Accepted: 5 November 2012 Ó The Author(s) 2012. This article is published with open access at Springerlink.com Abstract The decline of honeybees and other pollinating agricultural chemicals used to combat pests and fungi that insects is a current cause for concern. A major factor bees experience when they pollinate flowering crops or implicated in their decline is exposure to agricultural plants near agricultural land (Dainat et al. 2011; Neumann chemicals, in particular the neonicotinoid insecticides such and Carreck 2010; vanEngelsdorp et al. 2009). Systemic as imidacloprid. Honeybees are also subjected to additional insecticides, such as the neonicotinoids, are of particular chemical exposure when beekeepers treat hives with aca- concern as they persist in pollen and nectar long after ricides to combat the mite Varroa destructor. Here, we application (Rortais et al. 2005). Domesticated honeybees assess the effects of acute sublethal doses of the neoni- are also exposed to chemical acaricides administered by cotinoid imidacloprid, and the organophosphate acaricide beekeepers within the colony to control infestations of the coumaphos, on honey bee learning and memory. Imida- parasitic mite Varroa destructor (Rosenkranz et al. 2010). cloprid had little effect on performance in a six-trial It has been suggested that combined exposure to both olfactory conditioning assay, while coumaphos caused a pesticides and acaricides may be more toxic to bees than modest impairment. -

EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Aqueous Direct Injection and LC/MS/MS Summar
EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Aqueous Direct Injection and LC/MS/MS UCT Part Numbers: SLAQ100ID21-3UM - Selectra® Aqueous C18, 100 x 2.1mm, 3µm SLAQGDC20-3UM - Selectra® Aqueous C18, Guard column, 10 x 2.0mm, 3µm SLGRDHLDR - Guard Cartridge Holder June 2015 Summary: This application outlines a direct aqueous injection-liquid chromatography/tandem mass spectrometry (DAI-LC/MS/MS) method for the determination of 11 selected organic contaminants in drinking water, including methamidophos, acephate, aldicarb sulfoxide, oxydemeton methyl, dicrotophos, aldicarb, diisopropyl methylphosphonate (DIMP), fenamiphos sulfone, fenamiphos sulfoxide, thiofanox, and quinoline [1]. Dicrotophos, oxydemeton methyl, methamidophos, and acephate are UCMR4 compounds. An Aqueous C18 HPLC column was utilized for analyte retention and separation. Calibration curves were constructed using calibration standards prepared in reagent water with preservative reagents for analyte quantitation. The responses were linear over the entire analytical ranges (R2 ≥ 0.9970). Excellent accuracy (90 - 111%) and precision (RSD% < 20%, n=7) were achieved for fortified reagent water and tap water samples. Procedure: 1. Preserve drinking water sample with 64 mg/L of sodium omadine (antimicrobial) and 1.5 g/L of ammonium acetate (binding free chlorine). 2. Mix 0.99 mL of the preserved water sample with 10 μL of 0.4-12.5 ng/μL internal standard mixture, and vortex for 30 sec. 3. Inject 50 μL onto LC/MS/MS equipped with an aqueous -

Crystal Structures of Oxime-Bound Fenamiphos
Crystal structures of oxime-bound fenamiphos-acetylcholinesterases: reactivation involving flipping of the His447 ring to form a reactive Glu334-His447-Oxime triad Andreas Hörnberg, Elisabet Artursson, Rikard Wärme, Yuan-Ping Pang, Fredrik Ekström To cite this version: Andreas Hörnberg, Elisabet Artursson, Rikard Wärme, Yuan-Ping Pang, Fredrik Ekström. Crys- tal structures of oxime-bound fenamiphos-acetylcholinesterases: reactivation involving flipping of the His447 ring to form a reactive Glu334-His447-Oxime triad. Biochemical Pharmacology, Elsevier, 2009, 79 (3), pp.507. 10.1016/j.bcp.2009.08.027. hal-00538094 HAL Id: hal-00538094 https://hal.archives-ouvertes.fr/hal-00538094 Submitted on 21 Nov 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Accepted Manuscript Title: Crystal structures of oxime-bound fenamiphos-acetylcholinesterases: reactivation involving flipping of the His447 ring to form a reactive Glu334-His447-Oxime triad Authors: Andreas Hornberg,¨ Elisabet Artursson, Rikard Warme,¨ Yuan-Ping Pang, Fredrik Ekstrom¨ PII: S0006-2952(09)00730-8 -

Guide No. 1 – October 2020 2/12 the CONCEPT and IMPLEMENTATION of CPA GUIDANCE RESIDUE LEVELS
Cooperation Centre for Scientific Research Relative to Tobacco CORESTA GUIDE N° 1 The Concept and Implementation of CPA Guidance Residue Levels October 2020 Agro-Chemical Advisory Committee CORESTA TECHNICAL GUIDE N° 1 Title: The Concept and Implementation of CPA Guidance Residue Levels Status: Valid Note: This document will be periodically reviewed by CORESTA Document history: Date of review Information July 2003 Version 1 GRL for Pyrethrins () and Terbufos corrected. December 2003 CPA terminology corrected. June 2008 Version 2 – GRLs revised and residue definitions added Provisional GRL of 2.00 ppm for Cyfluthrin to replace previous June 2010 GRL of 0.50 ppm July 2013 Version 3 – GRLs revised October 2013 Note for Maleic Hydrazide revised Version 4 – GRLs revised + clarification that scope of GRLs July 2016 applies predominantly to the production of traditional cigarette tobaccos and GAP associated with their cultivation. June 2018 Fluopyram GRL of 5 ppm added to GRL list Version 5 – Nine new CPAs with GRL added to list. November 2019 Revision of GRLs for Chlorantraniliprole and Indoxacarb. Updated web links. October 2020 Version 6 – Flupyradifurone GRL of 21 ppm added to GRL list. CORESTA Guide No. 1 – October 2020 2/12 THE CONCEPT AND IMPLEMENTATION OF CPA GUIDANCE RESIDUE LEVELS Executive Summary • Guidance Residue Levels (GRLs) are in the remit of the Agro-Chemical Advisory Committee (ACAC) of CORESTA. Their development is a joint activity of all ACAC members, who represent the leaf production, processing and manufacturing sectors of the Tobacco Industry. The concept of GRLs and their implementation are described in this guide. • GRLs provide guidance to tobacco growers and assist with interpretation and evaluation of results from analyses of residues of Crop Protection Agents (CPAs*). -

Photocatalytic Degradation of Monocrotophos and Chlorpyrifos In
Journal of Water Process Engineering 7 (2015) 94–101 Contents lists available at ScienceDirect Journal of Water Process Engineering journa l homepage: www.elsevier.com/locate/jwpe Photocatalytic degradation of monocrotophos and chlorpyrifos in aqueous solution using TiO2 under UV radiation ∗ Augustine Amalraj, Anitha Pius Department of Chemistry, The Gandhigram Rural Institute – Deemed University, Gandhigram, Dindigul 624 302, Tamil Nadu, India a r t i c l e i n f o a b s t r a c t Article history: Monocrotophos (MCP) and chlorpyrifos (CPS) are most popular and broadly used organophosphorous Received 6 November 2014 pesticides owing to its low cost and high efficiency in controlling pests in agriculture. Presence of Received in revised form 2 June 2015 pesticides in aquatic environments causes serious problems to human beings and other organisms. Photo- Accepted 2 June 2015 catalytic degradation has been proved to be a promising method for the treatment of water. In view of this, Available online 17 June 2015 TiO2 photocatalyst was prepared by sol–gel method and characterized by SEM with EDAX, XRD, BET and FTIR. The photocatalytic degradation of MCP and CPS was carried out using prepared TiO2 photocatalyst Keywords: irradiated with 16 W UV light source. The effect of various parameters, i.e., photocatalyst concentration, TiO2 nanoparticles Pesticides pesticide concentration and pH of the solution on the percentage of degradation of selected pesticides Monocrotophos had been examined. The kinetic analysis of photodegradation of MCP and CPS under different initial con- Chlorpyrifos centration followed the Langmuir–Hinshelwood model. TiO2 found to be an excellent photocatalyst for Langmuir–Hinshelwood model the degradation of MCP and CPS under UV light irradiation. -
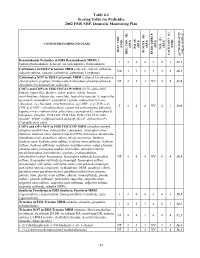
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime.