Crystal Structures of Oxime-Bound Fenamiphos
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water with Direct Aqueous Injection LC/MS/MS
EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water with Direct Aqueous Injection LC/MS/MS E. Michael Thurman and Imma Ferrer Center for Environmental Mass Spectrometry University of Colorado Boulder, CO, USA Confidentiality Label 1 March 20, 2012 Abstract EPA Method 538 is a new method from EPA for organophosphate pesticides in drinking water. It uses direct aqueous injection; thus, no sample preparation is needed. We use both UHPLC (Agilent 1290) and MS/MS (Agilent 6460) analysis for rapid analysis and sensitive detection with ng/L limits of detection. A second MRM is added for more reliable identification. Confidentiality Label 2 March 20, 2012 Hypothesis Direct injection of organophosphate pesticides (EPA Method 538) will work by UHPLC (Agilent Model 1290) and LC/MS/MS with Jetstream (Agilent Model 6460) with trace level detection at ng/L concentrations. Confidentiality Label 3 March 20, 2012 1. Introduction-Summary 1.1 EPA Method 538 (published in November 2009 by Shoemaker) deals with Organophosphate pesticides in drinking water (1) and one other contaminant, quinoline. 1.2 The method consists of 10 compounds: acephate, aldicarb, aldicarb sulfoxide, dicrotophos, diisopropylmethylphosphonate (DIMP), fenamiphos sulfone, fenamiphos sulfoxide, methamidophos, oxydemeton methyl, quinoline, and thiofanox with 5 labeled internal standards. 1.3 Direct aqueous injection is used with a large volume sample of 100 microliters; thus, no sample preparation is needed. 1.4 Because solid phase extraction (i.e. concentration of the sample is not carried out) suppression is mimimized in the analysis. 1.5 Part-per-Trillion Detection Limits. Confidentiality Label 4 March 20, 2012 Introduction 1.1: EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Direct Aqueous Injection by Jody Shoemaker, EPA Cincinnati, OH [email protected] 513-569-7298 Confidentiality Label 5 March 20, 2012 Introduction: 1.2. -

EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Aqueous Direct Injection and LC/MS/MS Summar
EPA Method 538: Determination of Selected Organic Contaminants in Drinking Water by Aqueous Direct Injection and LC/MS/MS UCT Part Numbers: SLAQ100ID21-3UM - Selectra® Aqueous C18, 100 x 2.1mm, 3µm SLAQGDC20-3UM - Selectra® Aqueous C18, Guard column, 10 x 2.0mm, 3µm SLGRDHLDR - Guard Cartridge Holder June 2015 Summary: This application outlines a direct aqueous injection-liquid chromatography/tandem mass spectrometry (DAI-LC/MS/MS) method for the determination of 11 selected organic contaminants in drinking water, including methamidophos, acephate, aldicarb sulfoxide, oxydemeton methyl, dicrotophos, aldicarb, diisopropyl methylphosphonate (DIMP), fenamiphos sulfone, fenamiphos sulfoxide, thiofanox, and quinoline [1]. Dicrotophos, oxydemeton methyl, methamidophos, and acephate are UCMR4 compounds. An Aqueous C18 HPLC column was utilized for analyte retention and separation. Calibration curves were constructed using calibration standards prepared in reagent water with preservative reagents for analyte quantitation. The responses were linear over the entire analytical ranges (R2 ≥ 0.9970). Excellent accuracy (90 - 111%) and precision (RSD% < 20%, n=7) were achieved for fortified reagent water and tap water samples. Procedure: 1. Preserve drinking water sample with 64 mg/L of sodium omadine (antimicrobial) and 1.5 g/L of ammonium acetate (binding free chlorine). 2. Mix 0.99 mL of the preserved water sample with 10 μL of 0.4-12.5 ng/μL internal standard mixture, and vortex for 30 sec. 3. Inject 50 μL onto LC/MS/MS equipped with an aqueous -

Guide No. 1 – October 2020 2/12 the CONCEPT and IMPLEMENTATION of CPA GUIDANCE RESIDUE LEVELS
Cooperation Centre for Scientific Research Relative to Tobacco CORESTA GUIDE N° 1 The Concept and Implementation of CPA Guidance Residue Levels October 2020 Agro-Chemical Advisory Committee CORESTA TECHNICAL GUIDE N° 1 Title: The Concept and Implementation of CPA Guidance Residue Levels Status: Valid Note: This document will be periodically reviewed by CORESTA Document history: Date of review Information July 2003 Version 1 GRL for Pyrethrins () and Terbufos corrected. December 2003 CPA terminology corrected. June 2008 Version 2 – GRLs revised and residue definitions added Provisional GRL of 2.00 ppm for Cyfluthrin to replace previous June 2010 GRL of 0.50 ppm July 2013 Version 3 – GRLs revised October 2013 Note for Maleic Hydrazide revised Version 4 – GRLs revised + clarification that scope of GRLs July 2016 applies predominantly to the production of traditional cigarette tobaccos and GAP associated with their cultivation. June 2018 Fluopyram GRL of 5 ppm added to GRL list Version 5 – Nine new CPAs with GRL added to list. November 2019 Revision of GRLs for Chlorantraniliprole and Indoxacarb. Updated web links. October 2020 Version 6 – Flupyradifurone GRL of 21 ppm added to GRL list. CORESTA Guide No. 1 – October 2020 2/12 THE CONCEPT AND IMPLEMENTATION OF CPA GUIDANCE RESIDUE LEVELS Executive Summary • Guidance Residue Levels (GRLs) are in the remit of the Agro-Chemical Advisory Committee (ACAC) of CORESTA. Their development is a joint activity of all ACAC members, who represent the leaf production, processing and manufacturing sectors of the Tobacco Industry. The concept of GRLs and their implementation are described in this guide. • GRLs provide guidance to tobacco growers and assist with interpretation and evaluation of results from analyses of residues of Crop Protection Agents (CPAs*). -

Lifetime Organophosphorous Insecticide Use Among Private Pesticide Applicators in the Agricultural Health Study
Journal of Exposure Science and Environmental Epidemiology (2012) 22, 584 -- 592 & 2012 Nature America, Inc. All rights reserved 1559-0631/12 www.nature.com/jes ORIGINAL ARTICLE Lifetime organophosphorous insecticide use among private pesticide applicators in the Agricultural Health Study Jane A. Hoppin1, Stuart Long2, David M. Umbach3, Jay H. Lubin4, Sarah E. Starks5, Fred Gerr5, Kent Thomas6, Cynthia J. Hines7, Scott Weichenthal8, Freya Kamel1, Stella Koutros9, Michael Alavanja9, Laura E. Beane Freeman9 and Dale P. Sandler1 Organophosphorous insecticides (OPs) are the most commonly used insecticides in US agriculture, but little information is available regarding specific OP use by individual farmers. We describe OP use for licensed private pesticide applicators from Iowa and North Carolina in the Agricultural Health Study (AHS) using lifetime pesticide use data from 701 randomly selected male participants collected at three time periods. Of 27 OPs studied, 20 were used by 41%. Overall, 95% had ever applied at least one OP. The median number of different OPs used was 4 (maximum ¼ 13). Malathion was the most commonly used OP (74%) followed by chlorpyrifos (54%). OP use declined over time. At the first interview (1993--1997), 68% of participants had applied OPs in the past year; by the last interview (2005--2007), only 42% had. Similarly, median annual application days of OPs declined from 13.5 to 6 days. Although OP use was common, the specific OPs used varied by state, time period, and individual. Much of the variability in OP use was associated with the choice of OP, rather than the frequency or duration of application. -
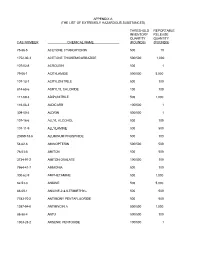
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Quantum Chemical Study of the Thermochemical Properties of Organophosphorous Compounds A
QUANTUM CHEMICAL STUDY OF THE THERMOCHEMICAL PROPERTIES OF ORGANOPHOSPHOROUS COMPOUNDS A. Khalfa, M. Ferrari, R. Fournet, B. Sirjean, L. Verdier, Pierre-Alexandre Glaude To cite this version: A. Khalfa, M. Ferrari, R. Fournet, B. Sirjean, L. Verdier, et al.. QUANTUM CHEMICAL STUDY OF THE THERMOCHEMICAL PROPERTIES OF ORGANOPHOSPHOROUS COMPOUNDS. Journal of Physical Chemistry A, American Chemical Society, 2015, 119 (42), pp.10527-10539. 10.1021/acs.jpca.5b07071. hal-01241498 HAL Id: hal-01241498 https://hal.archives-ouvertes.fr/hal-01241498 Submitted on 10 Dec 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. QUANTUM CHEMICAL STUDY OF THE THERMOCHEMICAL PROPERTIES OF ORGANOPHOSPHOROUS COMPOUNDS A. Khalfa, M. Ferrari1, R. Fournet1, B. Sirjean1, L. Verdier2, P.A. Glaude1 1Laboratoire Réactions et Génie des Procédés, Université de Lorraine, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France, 2DGA Maîtrise NRBC, Site du Bouchet, 5 rue Lavoisier, BP n°3, 91710 Vert le Petit, France Abstract Organophosphorous compounds are involved in many toxic compounds such as fungicides, pesticides, or chemical warfare nerve agents. The understanding of the decomposition chemistry of these compounds in the environment is largely limited by the scarcity of thermochemical data. -

Diazinon Use Summary
STATE OF WASHINGTON DEPARTMENT OF AGRICULTURE P.O. Box 42560 • Olympia, Washington 98504-2560 • (360) 902-1800 WASHINGTON STATE FENAMIPHOS USE SUMMARY • The products containing fenamiphos registered for use in Washington State are restricted use pesticides (RUP) and may be purchased and applied by only a certified applicator. • Fenamiphos is used to control a number of nematode pests. Nematodes can live as parasites on the outside or the inside of a plant. They may be free living or associated with cyst and root-knot formations in plants. • Fenamiphos is used on a wide variety of plants nationally including fruit, some vegetables, turf and grains. The compound is absorbed by the roots and translocated throughout the plant. • Fenamiphos blocks the enzyme acetylcholinesterase in the target pest. It also has secondary activity against other invertebrates such as sucking insects and spider mites. • Fenamiphos is available in emulsifiable concentrate, granular or emulsion formulations. It may also be found in formulations with other pesticides such as carbofuran and disulfoton. • Fenamiphos is classified toxicity class I - highly toxic. Products containing fenamiphos bear the signal word, “Danger.” Fenamiphos belongs to the organophosphate chemical class. • Fenamiphos is labeled for use on several crops in Washington State. (See table, “Product Names and Labeled Crops” provided below.) With the exception of red raspberries, it is not commonly used. WASS* EST. % EST. EST. LBS. 2001 EST. EST. LBS. # OF ACRES ACRES A.I. CROP ACRES A.I./ACRE APPS TREATED TREATED APPLIED PLANTED Red raspberries 9,500 10 3.0 1 950 2850 * Washington Agricultural Statistics Service Washington State Department of Agriculture/Endangered Species Program Page 1 of 4 http://agr.wa.gov/PestFert/EnvResources/EndangSpecies.htm November 2003 MAJOR USES (listed alphabetically): The major use listing supplies the most commonly used formulations of the active ingredient. -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

Fenamiphos Proposed AEGL Document
ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) FOR FENAMIPHOS CAS Reg. No. 22224-92-6 PROPOSED FENAMIPHOS PROPOSED: November 2009 1 2 ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 3 FOR 4 FENAMIPHOS 5 (CAS Reg. No. 22224-92-6) 6 7 8 9 10 PROPOSED 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 2 FENAMIPHOS PROPOSED: November 2009 1 2 3 PREFACE 4 5 Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 6 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous 7 Substances (NAC/AEGL Committee) has been established to identify, review and interpret 8 relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic 9 chemicals. 10 11 AEGLs represent threshold exposure limits for the general public and are applicable to 12 emergency exposure periods ranging from 10 minutes to 8 hours. Three levels C AEGL-1, 13 AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 14 hour, 4 hours, and 8 hours) and are distinguished by varying degrees of severity of toxic effects. 15 The three AEGLs are defined as follows: 16 17 AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per 18 cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general 19 population, including susceptible individuals, could experience notable discomfort, irritation, or 20 certain asymptomatic, non-sensory effects. However, the effects are not disabling and are 21 transient and reversible upon cessation of exposure. -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation.