Toward On-Demand Profile Hidden Markov Models for Genetic Barcode Identification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

( ) Hydropsychidae (Insecta: Trichoptera) As Bio-Indicators Of
ว.วิทย. มข. 40(3) 654-666 (2555) KKU Sci. J. 40(3) 654-666 (2012) แมลงน้ําวงศ!ไฮดรอบไซคิดี้ (อันดับไทรคอบเทอร-า) เพื่อเป2นตัวบ-งชี้ทางชีวภาพของคุณภาพน้ํา Hydropsychidae (Insecta: Trichoptera) as Bio-indicators of Water QuaLity แตงออน พรหมมิ1 บทคัดยอ การประเมินคุณภาพน้ําในแมน้ําและลําธารควรที่จะมีการใชปจจัยทางกายภาพ เคมีและชีวภาพควบคูกัน ไป ปจจัยทางชีวภาพที่มีศักยภาพในการประเมินคุณภาพน้ําในแหลงน้ําคือกลุมสัตว+ไมมีกระดูกสันหลังขนาดใหญที่ อาศัยอยูตามพื้นทองน้ํา โดยเฉพาะแมลงน้ําอันดับไทรคอบเทอรา ซึ่งเป3นกลุมสัตว+ที่มีความหลากหลายมากกลุม หนึ่งในแหลงน้ํา ระยะตัวออนของแมลงกลุมนี้ทุกชนิดอาศัยอยูในแหลงน้ํา เป3นองค+ประกอบหลักในแหลงน้ําและ เป3นตัวหมุนเวียนสารอาหารในแหลงน้ํา ระยะตัวออนของแมลงน้ํากลุมนี้จะตอบสนองตอปจจัยของสภาพแวดลอม ในแหลงน้ําทุกรูปแบบ ระยะตัวเต็มวัยอาศัยอยูบนบกบริเวณตนไมซึ่งไมไกลจากแหลงน้ํามากนัก หากินเวลา กลางคืน ความรูทางดานอนุกรมวิธานและชีววิทยาไมวาจะเป3นระยะตัวออนหรือตัวเต็มวัยของแมลงน้ําอันดับไทร คอบเทอราในประเทศแถบยุโรปตะวันตกและอเมริกาเหนือสามารถวินิจฉัยไดถึงระดับชนิด โดยเฉพาะแมลงน้ํา วงศ+ไฮดรอบไซคิดี้ มีการประยุกต+ใชในการติดตามตรวจสอบทางชีวภาพของคุณภาพน้ํา เนื่องจากชนิดของตัวออน แมลงน้ําวงศ+นี้มีความทนทานตอมลพิษในชวงกวางมากกวาแมลงน้ําชนิดอื่น ๆ 1สายวิชาวิทยาศาสตร+ คณะศิลปศาสตร+และวิทยาศาสตร+ มหาวิทยาลัยเกษตรศาสตร+ วิทยาเขตกําแพงแสน จ.นครปฐม 73140 E-mail: [email protected] บทความ วารสารวิทยาศาสตร+ มข. ปQที่ 40 ฉบับที่ 3 655 ABSTRACT Assessment on rivers and streams water quality should incorporate aspects of chemical, physical, and biological. Of all the potential groups of freshwater organisms that have been considered for -

Trichoptera:Hydropsychidae) Based on DNA and Morphological Evidence Christy Jo Geraci National Museum on Natural History, Smithsonian Institute
Clemson University TigerPrints Publications Biological Sciences 3-2010 Defining the Genus Hydropsyche (Trichoptera:Hydropsychidae) Based on DNA and Morphological Evidence Christy Jo Geraci National Museum on Natural History, Smithsonian Institute Xin Zhou University of Guelph John C. Morse Clemson University, [email protected] Karl M. Kjer Rutgers University - New Brunswick/Piscataway Follow this and additional works at: https://tigerprints.clemson.edu/bio_pubs Part of the Biology Commons Recommended Citation Please use publisher's recommended citation. This Article is brought to you for free and open access by the Biological Sciences at TigerPrints. It has been accepted for inclusion in Publications by an authorized administrator of TigerPrints. For more information, please contact [email protected]. J. N. Am. Benthol. Soc., 2010, 29(3):918–933 ’ 2010 by The North American Benthological Society DOI: 10.1899/09-031.1 Published online: 29 June 2010 Defining the genus Hydropsyche (Trichoptera:Hydropsychidae) based on DNA and morphological evidence Christy Jo Geraci1 Department of Entomology, National Museum of Natural History, Smithsonian Institution, Washington, DC 20013-7012 USA Xin Zhou2 Biodiversity Institute of Ontario, University of Guelph, Guelph, Ontario, N1G 2W1 Canada John C. Morse3 Department of Entomology, Soils, and Plant Sciences, Clemson University, Clemson, South Carolina 29634 USA Karl M. Kjer4 Department of Ecology, Evolution and Natural Resources, School of Environmental and Biological Sciences, Rutgers University, New Brunswick, New Jersey 08901 USA Abstract. In this paper, we review the history of Hydropsychinae genus-level classification and nomenclature and present new molecular evidence from mitochondrial cytochrome c oxidase subunit I (COI) and nuclear large subunit ribosomal ribonucleic acid (28S) markers supporting the monophyly of the genus Hydropsyche. -
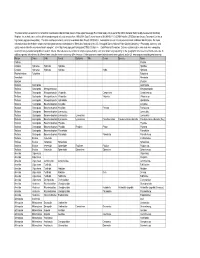
This Table Contains a Taxonomic List of Benthic Invertebrates Collected from Streams in the Upper Mississippi River Basin Study
This table contains a taxonomic list of benthic invertebrates collected from streams in the Upper Mississippi River Basin study unit as part of the USGS National Water Quality Assessemnt (NAWQA) Program. Invertebrates were collected from woody snags in selected streams from 1996-2004. Data Retreival occurred 26-JAN-06 11.10.25 AM from the USGS data warehouse (Taxonomic List Invert http://water.usgs.gov/nawqa/data). The data warehouse currently contains invertebrate data through 09/30/2002. Invertebrate taxa can include provisional and conditional identifications. For more information about invertebrate sample processing and taxonomic standards see, "Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory -- Processing, taxonomy, and quality control of benthic macroinvertebrate samples", at << http://nwql.usgs.gov/Public/pubs/OFR00-212.html >>. Data Retrieval Precaution: Extreme caution must be exercised when comparing taxonomic lists generated using different search criteria. This is because the number of samples represented by each taxa list will vary depending on the geographic criteria selected for the retrievals. In addition, species lists retrieved at different times using the same criteria may differ because: (1) the taxonomic nomenclature (names) were updated, and/or (2) new samples containing new taxa may Phylum Class Order Family Subfamily Tribe Genus Species Taxon Porifera Porifera Cnidaria Hydrozoa Hydroida Hydridae Hydridae Cnidaria Hydrozoa Hydroida Hydridae Hydra Hydra sp. Platyhelminthes Turbellaria Turbellaria Nematoda Nematoda Bryozoa Bryozoa Mollusca Gastropoda Gastropoda Mollusca Gastropoda Mesogastropoda Mesogastropoda Mollusca Gastropoda Mesogastropoda Viviparidae Campeloma Campeloma sp. Mollusca Gastropoda Mesogastropoda Viviparidae Viviparus Viviparus sp. Mollusca Gastropoda Mesogastropoda Hydrobiidae Hydrobiidae Mollusca Gastropoda Basommatophora Ancylidae Ancylidae Mollusca Gastropoda Basommatophora Ancylidae Ferrissia Ferrissia sp. -

A Revision of the Lotic Genus Potamyia BANKS 1900 (Trichoptera: Hydropsychidae) with the Description of Eight New Species
© Biologiezentrum Linz/Austria; download unter www.biologiezentrum.at Linzer biol. Beitr. 38/1 739-777 21.7.2006 A revision of the lotic genus Potamyia BANKS 1900 (Trichoptera: Hydropsychidae) with the description of eight new species J. OLÁH, P.C. BARNARD & H. MALICKY A b s t r a c t : The taxonomic position, ecology and distribution of the lotic genus Potamyia BANKS are revised. The previous distribution of the genus in N. America and the Oriental region is greatly extended, with six species discovered in Madagascar. Of the 42 species now recognised, eight are described as new: P. ifanadiana nov.spec., P. tofina nov.spec., P. andoba nov.spec., P. karafa nov.spec., P. andria nov.spec., P. korasha nov.spec., P. nuonga nov.spec., P. trenhona nov.spec. Five species are transferred to the genus: P. chekiangensis (SCHMID), P. dentifera (ULMER), P. pallidipennis (MARTYNOV), P. hoenei (SCHMID), P. nikalandugola (SCHMID). Five new species synonymies are established. K e y w o r d s : Trichoptera, Hydropsychidae, Potamyia, Taxonomy, Madagascar, Oriental, new species. Introduction Potamyia was erected by BANKS (1900) as a new hydropsychid genus near to Macronema, with Macronema flavum HAGEN 1861, a species living in large North American rivers, as the type-species. ULMER (1912) described a fossil species, P. nitida from Baltic amber, but according to MARTYNOV (1930) this Eocene hydropsychid probably belongs in Hydropsyche. The only other species known in the genus until re- cently were P. chinensis (ULMER), P. trilobata (ULMER), here recognised as a synonym of P. straminea (MCLACHLAN), and P. yunnanica (SCHMID), all known from China, together with Cheumatopsyche czekanovskii MARTYNOV known as an abundant potamo- biont in the large Siberian rivers (recorded from Mongolia and transferred to Potamyia by SCHMID 1967, with females recorded from Korea by BOTOSANEANU (1970). -

Refinement of the Basin-Wide Index of Biotic Integrity for Non-Tidal Streams and Wadeable Rivers in the Chesapeake Bay Watershed
Refinement of the Basin-Wide Index of Biotic Integrity for Non-Tidal Streams and Wadeable Rivers in the Chesapeake Bay Watershed APPENDICES Appendix A: Taxonomic Classification Appendix B: Taxonomic Attributes Appendix C: Taxonomic Standardization Appendix D: Rarefaction Appendix E: Biological Metric Descriptions Appendix F: Abiotic Parameters for Evaluating Stream Environment Appendix G: Stream Classification Appendix H: HUC12 Watershed Characteristics in Bioregions Appendix I: Index Methodologies Appendix J: Scoring Methodologies Appendix K: Index Performance, Accuracy, and Precision Appendix L: Narrative Ratings and Maps of Index Scores Appendix M: Potential Biases in the Regional Index Ratings Appendix Citations Appendix A: Taxonomic Classification All taxa reported in Chessie BIBI database were assigned the appropriate Phylum, Subphylum, Class, Subclass, Order, Suborder, Family, Subfamily, Tribe, and Genus when applicable. A portion of the taxa reported were reported under an invalid name according to the ITIS database. These taxa were subsequently changed to the taxonomic name deemed valid by ITIS. Table A-1. The taxonomic hierarchy of stream macroinvertebrate taxa included in the Chesapeake Bay non-tidal database. -

Redalyc.Especies Del Orden Thichoptera (Insecta) En Colombia
Biota Colombiana ISSN: 0124-5376 [email protected] Instituto de Investigación de Recursos Biológicos "Alexander von Humboldt" Colombia Muñoz Quesada, Fernando Especies del Orden Thichoptera (Insecta) en Colombia Biota Colombiana, vol. 1, núm. 3, diciembre, 2000, pp. 267-288 Instituto de Investigación de Recursos Biológicos "Alexander von Humboldt" Bogotá, Colombia Disponible en: http://www.redalyc.org/articulo.oa?id=49110304 Cómo citar el artículo Número completo Sistema de Información Científica Más información del artículo Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Página de la revista en redalyc.org Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto VarónBiota Colombiana 1 (3) 267 - 288, 2000 Eumastacids of Colombia - 267 Especies del Orden Trichoptera (Insecta) en Colombia Fernando Muñoz-Quesada Department of Entomology, University of Minnesota, 1980 Folwell Ave., St.Paul, MN 55108, U.S.A. [email protected]; [email protected] Palabras Clave: Trichoptera, Insectos Acuáticos, Listado de Especies, Colombia, Neotrópico Trichoptera es el orden de insectos cuyas alas, princi- Trichoptera poseen además de los tres pares de patas palmente las anteriores, están cubiertas por pelos (del grie- torácicas, un solo par de propatas anales con una uña en el go “trichos” = pelos, “pteron” = ala, “ala con pelos”), y último segmento abdominal, y no presentan espiráculos que presentan metamorfosis completa (holometábolos). Las abiertos (apneústicas). Las larvas tienen una glándula formas inmaduras (huevo, larva y pupa) son generalmente salival modificada que les permite producir seda para sos- acuáticas y se encuentran comúnmente en ríos, riachuelos, tenerse en el sustrato o para construir redes para filtrar cascadas y lagos. -

DBR Y W OREGON STATE
The Distribution and Biology of the A. 15 Oregon Trichoptera PEE .1l(-.", DBR Y w OREGON STATE Technical Bulletin 134 AGRICULTURAL 11 EXPERIMENTI STATION Oregon State University Corvallis, Oregon INovember 1976 FOREWORD There are four major groups of insectswhoseimmature stages are almost all aquatic: the caddisflies (Trichoptera), the dragonflies and damselflies (Odonata), the mayflies (Ephemeroptera), and the stoneflies (Plecoptera). These groups are conspicuous and important elements in most freshwater habitats. There are about 7,000 described species of caddisflies known from the world, and about 1,200 of these are found in America north of Mexico. All play a significant ro'e in various aquatic ecosystems, some as carnivores and others as consumers of plant tissues. The latter group of species is an important converter of plant to animal biomass. Both groups provide food for fish, not only in larval but in pupal and adult stages as well. Experienced fishermen have long imitated these larvae and adults with a wide variety of flies and other artificial lures. It is not surprising, then, that the caddisflies have been studied in detail in many parts of the world, and Oregon, with its wide variety of aquatic habitats, is no exception. Any significant accumulation of these insects, including their various develop- mental stages (egg, larva, pupa, adult) requires the combined efforts of many people. Some collect, some describe new species or various life stages, and others concentrate on studying and describing the habits of one or more species. Gradually, a body of information accumulates about a group of insects for a particular region, but this information is often widely scattered and much effort is required to synthesize and collate the knowledge. -

New Species of Plectropsyche Ross 1947 (Trichoptera: Hydropsychidae: Hydropsychinae)
Zootaxa 4040 (4): 421–432 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2015 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.4040.4.2 http://zoobank.org/urn:lsid:zoobank.org:pub:B50C1290-A6F7-461E-AC0F-F490F26E9780 New species of Plectropsyche Ross 1947 (Trichoptera: Hydropsychidae: Hydropsychinae) JOAQUÍN BUENO-SORIA¹ & RAFAEL BARBA-ALVAREZ² ¹141 Preamble Dr. Marlton, NJ 08053-5350, USA. E-mail: [email protected] ²Instituto de Biología, Departamento de Zoología UNAM. AP 70-153, Ciudad Universitaria, 3erCto. Exterior S/N. México 04510 D.F. E-mail: [email protected] Abstract Two new species from Mexico and Central America of the genus Plectropsyche Ross are described and the male and fe- male genitalia are figured. The only extant previous described species, Plectropsyche hoogstraali Ross and its synonym Plectropsyche pitella (Denning) from Mexico are studied. A map with the distribution of all known Plectropsyche species is presented and an illustrated key for the males of the extant species are included. Key words: taxonomy, Mexico, Central America Resumen Se describen dos especies nuevas del género Plectropsyche Ross de México y Centroamérica y se ilustran los genitalia de los machos y las hembras. Unicamente se estudiaron las especies actuales previamente descritas para México, Plectrop- syche hoogstraali Ross y su sinónimo P. pitella (Denning). Se incluye un mapa con la distribución de las especies así como una clave ilustrada de los machos del género. Introduction The genus Plectropsyche Ross 1947, with type species Plectropsyche hoogstraali Ross 1947, is considered to be closely related to the genera Cheumatopsyche Wallengren 1891 and Potamyia Banks 1900. -

Descriptions of Larvae of Four Species of Hydropsyche (Hydropsychidae: Trichoptera) from Thailand
Zootaxa 4158 (4): 577–591 ISSN 1175-5326 (print edition) http://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2016 Magnolia Press ISSN 1175-5334 (online edition) http://doi.org/10.11646/zootaxa.4158.4.9 http://zoobank.org/urn:lsid:zoobank.org:pub:D4CD004C-99EF-46CC-AF7F-9702DF55694D Descriptions of larvae of four species of Hydropsyche (Hydropsychidae: Trichoptera) from Thailand TAENG ON PROMMI Faculty of Liberal Arts and Science, Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom, 73140, THAILAND E-mail mail: [email protected] Abstract Morphological characters of the larvae of four species in the genus Hydropsyche (H. dolosa, H. brontes, H. napaea, and H. camillus) are described and figured for the first time. The most important diagnostic features are illustrated, and some information regarding their distribution, biology, and habitat are included. Key words: morphology, Hydropsyche dolosa, Hydropsyche brontes, Hydropsyche camillus, Hydropsyche napaea Introduction Species-level identification of aquatic insects is often required for ecological, evolutionary, or biomonitoring studies. Identification can be difficult because many formal descriptions are based only on adult males, which are often the most morphologically variant sex and life stage (Zhou et al. 2010). Caddisfly larvae (Trichoptera) are an important component of the benthic fauna in many streams and are routinely used in environmental assessments because of the relatively high sensitivity of most caddisfly species to environmental change. For many species, accurate identification of larvae requires the collection and study of the terrestrial adults and association of those identifiable adults with the yet-unidentifiable larvae. The knowledge of taxonomy of Thai caddisflies has been confined mainly to descriptions of the adult stages (Malicky 2010). -

The Caddisflies (Insecta: Trichoptera) of the Lake Itasca Region, Minnesota, and a Preliminary Assessment of the Conservation Status of Minnesota Trichoptera
Conservation Biology Research Grants Program Nongame Wildlife Program Division of Ecological Services Minnesota Department of Natural Resources THE CADDISFLIES (INSECTA: TRICHOPTERA) OF THE LAKE ITASCA REGION, MINNESOTA, AND A PRELIMINARY ASSESSMENT OF THE CONSERVATION STATUS OF MINNESOTA TRICHOPTERA A THESIS SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY MARGOT PECK MONSON IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER MASTER OF SCIENCE SEPTEMBER 1994 1 © Margot Peck Monson 1994 2 ACKNOWLEDGMENTS I would like to express my sincere gratitude to my major advisor, Dr. Ralph Holzenthal, for taking me on as his student and for his time, effort, and patient guidance in helping me pursue this project. I also wish to thank Dr. Roger Moon for his helpful counsel and encouragement, as well as the other members of my examination committee, Dr. William Miller and Dr. Francesca Cuthbert, for their support. I am grateful to Dr. O. J. Flint, Dr. Steve Harris, Dr. David Etnier, and the Illinois Natural History Survey for their assistance in making specimens available and for verification of some determinations. Dr. Etnier was also most generous in making several donations to the University of Minnesota Insect Collection. I am thankful for the partial funding for this project, contributed by the Nongame Wildlife Program of the Minnesota Department of Natural Resources and the Minnesota Chapter of The Nature Conservancy. The helpfulness of Dr. Phil Clausen, Curator of the University of Minnesota Insect Collection, was most appreciated (as was his willingness to answer my many questions). The friendship and good humor extended by my colleagues in the lab, Roger, Atilano, Jolanda, Sonia, John, and Marc, as well as Sue, Paul, Sujaya, and Diann will be fondly remembered. -

NATURAL HISTORY of THREE HYDROPSYCHIDAE (TRICHOPTERA, INSECTA) Ln a "CERRADO" STREAM from NORTHEASTERN SÃO PAULO, BRAZIL
NATURAL HISTORY OF THREE HYDROPSYCHIDAE (TRICHOPTERA, INSECTA) lN A "CERRADO" STREAM FROM NORTHEASTERN SÃO PAULO, BRAZIL Leandro Gonçalves Oliveira 1 Claudio Gilberto Froehlich 2 ABSTRACT. The tàunal composition 01' lhe Hydropsychidae (Trichoptera) 01' Pedregulho Stream is presented. Feeding habits of larvae and behavioural aspects of both larvae and adults are described. KEY WORDS. Trichoptera, Hydropsychidae, SlIIicridea, Leplonema, biology, larvae, bel1aviour Biological studies on the Brazilian aquatic insect fauna are stil\ incipient. This is particular\y valid for lhe Trichoplera. Some references on the subject are MÜLLER (1880), SCHUBART (1946), V ANZOUNI (1964), SCHROEDER-ARAUJO & C1POLU (1986), OLIVEIRA (1988, 1991) and NESSIMIAN (1995). A general presentation on lhe Trichoptera col\ected in Córrego do Pe dregulho, São Paulo, and a discussion of lhe role of abiotic factors is found in OLIVEIRA & FROEHUCH (in press). ln this paper aspects of the natural history of Hydropsychidae, in particular of lhe genera Smicridea and Leptonema, are pre sented. The cosmopolitan Hydropsychidae is one oflhe largest Trichoptera families in running waters (HAUER & STANFORD 1981; DEUTSCH 1984). Most larvae are col leclors-filterers (WIGGINS 1977; MACKA Y 1984); they can be recognized by the numerous abdominal gil\s and by the sclerotized thoracic terga (MERRlTT & CUMMlNS 1979; FLINT 1982; ROLDÁ N 1990). They build fixed tubular retreats with small stones and plant fragments, wilh the opening directed towards the current (WIGGlNS 1977). Smicridea Mclachlan, 1871, is a very common hydropsychid genus and the sole representative of the subfamily Hydropsychinae in South America (FLINT 1974). lt has been the subject oftaxonomic studies in Argentina (FUNT 1980, 1982, 1983), Chile (FLlNT 1989) and Brazil (FUNT 1978, 1983). -

Trichopterological Literature 45-53 © Hans Malicky/Austria; Download Unter 45
ZOBODAT - www.zobodat.at Zoologisch-Botanische Datenbank/Zoological-Botanical Database Digitale Literatur/Digital Literature Zeitschrift/Journal: Braueria Jahr/Year: 2006 Band/Volume: 33 Autor(en)/Author(s): Malicky Hans Artikel/Article: Trichopterological literature 45-53 © Hans Malicky/Austria; download unter www.biologiezentrum.at 45 Trichopterological literature Bello.C.L, Alba-Tercedor.J. 2004 Efecto de la regulacion de la cabecera del rio Genii (Sierra Nevada, 1992 Espana) sobre la comunidad de macroinvertebrados acuaticos y la dieta larvaria de Rhyacophila nevada (Insecta: Trichoptera). - de Moor, F.C. 1992 Limnetica 23:361-370. Parasites, generalist and specialist predators and their role in limiting the population size of blackflies and in particular Simulium Bispo.Pitagoras C, Oliveira.Leandro G., Crisci-Bispo.Vera L. 2004 chutteri Lewis (Diptera: Simuliidae) in and along the Vaal River, Environmental factors influencing distribution and abundance of South Africa. - Ann.Cape Prov.Mus.(nat.Hist) 18:271-291. trichopteran larvae in central Brazilian mountain streams. - Studies on neotropical Fauna and Environment 39:233-237. 1999 Botosaneanu.L; Thomas.A. 2004 Loewen.Mark A., Leggitt,V.Leroy, Buchheim.H.Paul 1999 Nouvelles contributions à la connaissance des Trichoptères de Caddisfly (Trichoptera) larval cases from Eocene Fossil lake, Fossil Martinique, avec description de deux espèces nouvelles Butte National Monument, pp. 72-77. In: Santucci.Vincent L, (Trichoptera). - Ephemera 6:33-58. McClelland.Lindsay (eds.): National Park Service Paleontol. Res. 4 (US NatPark Service, Geol.Res.Div.,Lakewood) Botosaneanu.Lazare, de Vos.Rob 2004 Two new species of Anisocentropus McLachlan, 1863 from 2000 Indonesian New Guinea (Papua) (Trichoptera: Calamoçeratidae), and a contribution to the knowledge of a third one.