Evolutionary Genomics in Corvids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alalā Or Hawaiian Crow (Corvus Hawaiiensis) 5-Year
`Alalā or Hawaiian Crow (Corvus hawaiiensis) 5-Year Review Summary and Evaluation U.S. Fish and Wildlife Service Pacific Islands Fish and Wildlife Office Honolulu, Hawai`i 5-YEAR REVIEW Species reviewed: `Alalā or Hawaiian Crow (Corvus hawaiiensis) TABLE OF CONTENTS 1.0 GENERAL INFORMATION .......................................................................................... 1 1.1 Reviewers ....................................................................................................................... 1 1.2 Methodology used to complete the review: ................................................................. 1 1.3 Background ................................................................................................................... 1 2.0 REVIEW ANALYSIS ....................................................................................................... 2 2.1 Application of the 1996 Distinct Population Segment (DPS) policy ......................... 2 2.2 Recovery Criteria .......................................................................................................... 3 2.3 Updated Information and Current Species Status .................................................... 4 2.4 Synthesis......................................................................................................................... 9 3.0 RESULTS .......................................................................................................................... 9 3.1 Recommended Classification ...................................................................................... -

Japan in Winter January 21–February 2, 2017
JAPAN IN WINTER JANUARY 21–FEBRUARY 2, 2017 LEADERS: KAZ SHINODA & BOB SUNDSTROM WITH KOJI NIIYA ON HOKKAIDO LIST COMPILED BY: BOB SUNDSTROM VICTOR EMANUEL NATURE TOURS, INC. 2525 WALLINGWOOD DRIVE, SUITE 1003 AUSTIN, TEXAS 78746 WWW.VENTBIRD.COM JAPAN IN WINTER: A CRANE & SEA-EAGLE SPECTACLE! By Bob Sundstrom The Japan in Winter tour is subtitled “A Crane and Sea-Eagle Spectacle,” a billing it truly lives up to. The tour has been designed by Japanese birding tour leader, Kaz Shinoda, and traverses the three main—and distinctively different—islands of Honshu, Kyushu, and Hokkaido. About a third of the tour is devoted to each island. From south to north, that’s a span of over 1,600 miles, from the mild weather and green tea plantations of Kyushu to the snowy rim of the Sea of Okhotsk on Hokkaido, with the largest, central island of Honshu in-between. On our 2017 tour we enjoyed great views of five species of cranes. At Arasaki on the island of Kyushu, we took in one of the grandest crane spectacles in the world as thousands of White- naped and Hooded cranes gathered in fields and paddies: tall, silvery White-naped Cranes with bold red faces standing head and shoulders above petite, elegant Hooded Cranes with charcoal bodies, white necks, and red forecrowns. Tucked among their thousands were a handful of Eurasian/Common Cranes and Sandhill Cranes. At sunset, most of the vast assemblage flew by as we watched, a few dozen or a few hundred at a time, with the orange sky of late sunset behind them as they winged to a nighttime roost nearby. -

Mongolia's Snow Leopards
Mongolia’s Snow Leopards Naturetrek Tour Report 27 August - 7 September 2019 Cossac Fox by Adam Dudley Snow Leopard by Gerald Broddelez Dione Snake by Gerald Broddelez Przewalski's Horse by Jane Dixon Report compiled by Gerald Broddelez Images courtesy of Jane Dixon, Adam Dudley & Gerald Broddelez Naturetrek Mingledown Barn Wolf's Lane Chawton Alton Hampshire GU34 3HJ UK T: +44 (0)1962 733051 E: [email protected] W: www.naturetrek.co.uk Tour Report Mongolia’s Snow Leopards Tour participants: Gerald Broddelez (leader), Terbish and Sovd (local guides) with 12 Naturetrek clients Day 1 Tuesday 27th August The group was in flight via Moscow to Ulaanbaatar (known as UB). Day 2 Wednesday 28th August The plane arrived with some delay, so our plans for the morning were rearranged accordingly. We drove to our hotel for the night and enjoyed some free time before an early lunch. During the afternoon we did some birding in the river area and found a good selection of birds that included several groups of smart Azure Tits, a large group of Azure-winged Magpies, several tristis forms of Chifchaff, a single Dusky Warbler, Daurian Jackdaw, many Taiga Flycatchers, Lesser Spotted Woodpecker and loads of raptors. Most were Kites but also Cinereous Vultures, a single Upland Buzzard, a female Goshawk, a close light- phase Booted Eagle and a Hobby! Several butterflies were flitting around in the warm air and included Small and Large White, Small Tortoiseshell and Painted Lady. Day 3 Thursday 29th August A Long-eared Owl was calling during the night and was seen by a few people in the spotlight. -

Federal Register/Vol. 85, No. 74/Thursday, April 16, 2020/Rules
21282 Federal Register / Vol. 85, No. 74 / Thursday, April 16, 2020 / Rules and Regulations DEPARTMENT OF THE INTERIOR United States and the Government of United States or U.S. territories as a Canada Amending the 1916 Convention result of recent taxonomic changes; Fish and Wildlife Service between the United Kingdom and the (8) Change the common (English) United States of America for the names of 43 species to conform to 50 CFR Part 10 Protection of Migratory Birds, Sen. accepted use; and (9) Change the scientific names of 135 [Docket No. FWS–HQ–MB–2018–0047; Treaty Doc. 104–28 (December 14, FXMB 12320900000//201//FF09M29000] 1995); species to conform to accepted use. (2) Mexico: Convention between the The List of Migratory Birds (50 CFR RIN 1018–BC67 United States and Mexico for the 10.13) was last revised on November 1, Protection of Migratory Birds and Game 2013 (78 FR 65844). The amendments in General Provisions; Revised List of this rule were necessitated by nine Migratory Birds Mammals, February 7, 1936, 50 Stat. 1311 (T.S. No. 912), as amended by published supplements to the 7th (1998) AGENCY: Fish and Wildlife Service, Protocol with Mexico amending edition of the American Ornithologists’ Interior. Convention for Protection of Migratory Union (AOU, now recognized as the American Ornithological Society (AOS)) ACTION: Final rule. Birds and Game Mammals, Sen. Treaty Doc. 105–26 (May 5, 1997); Check-list of North American Birds (AOU 2011, AOU 2012, AOU 2013, SUMMARY: We, the U.S. Fish and (3) Japan: Convention between the AOU 2014, AOU 2015, AOU 2016, AOS Wildlife Service (Service), revise the Government of the United States of 2017, AOS 2018, and AOS 2019) and List of Migratory Birds protected by the America and the Government of Japan the 2017 publication of the Clements Migratory Bird Treaty Act (MBTA) by for the Protection of Migratory Birds and Checklist of Birds of the World both adding and removing species. -

Appendix, French Names, Supplement
685 APPENDIX Part 1. Speciesreported from the A.O.U. Check-list area with insufficient evidencefor placementon the main list. Specieson this list havebeen reported (published) as occurring in the geographicarea coveredby this Check-list.However, their occurrenceis considered hypotheticalfor one of more of the following reasons: 1. Physicalevidence for their presence(e.g., specimen,photograph, video-tape, audio- recording)is lacking,of disputedorigin, or unknown.See the Prefacefor furtherdiscussion. 2. The naturaloccurrence (unrestrained by humans)of the speciesis disputed. 3. An introducedpopulation has failed to becomeestablished. 4. Inclusionin previouseditions of the Check-listwas basedexclusively on recordsfrom Greenland, which is now outside the A.O.U. Check-list area. Phoebastria irrorata (Salvin). Waved Albatross. Diornedeairrorata Salvin, 1883, Proc. Zool. Soc. London, p. 430. (Callao Bay, Peru.) This speciesbreeds on Hood Island in the Galapagosand on Isla de la Plata off Ecuador, and rangesat seaalong the coastsof Ecuadorand Peru. A specimenwas takenjust outside the North American area at Octavia Rocks, Colombia, near the Panama-Colombiaboundary (8 March 1941, R. C. Murphy). There are sight reportsfrom Panama,west of Pitias Bay, Dari6n, 26 February1941 (Ridgely 1976), and southwestof the Pearl Islands,27 September 1964. Also known as GalapagosAlbatross. ThalassarchechrysosWma (Forster). Gray-headed Albatross. Diornedeachrysostorna J. R. Forster,1785, M6m. Math. Phys. Acad. Sci. Paris 10: 571, pl. 14. (voisinagedu cerclepolaire antarctique & dansl'Ocean Pacifique= Isla de los Estados[= StatenIsland], off Tierra del Fuego.) This speciesbreeds on islandsoff CapeHorn, in the SouthAtlantic, in the southernIndian Ocean,and off New Zealand.Reports from Oregon(mouth of the ColumbiaRiver), California (coastnear Golden Gate), and Panama(Bay of Chiriqu0 are unsatisfactory(see A.O.U. -

Gear for a Big Year
APPENDIX 1 GEAR FOR A BIG YEAR 40-liter REI Vagabond Tour 40 Two passports Travel Pack Wallet Tumi luggage tag Two notebooks Leica 10x42 Ultravid HD-Plus Two Sharpie pens binoculars Oakley sunglasses Leica 65 mm Televid spotting scope with tripod Fossil watch Leica V-Lux camera Asics GEL-Enduro 7 trail running shoes GoPro Hero3 video camera with selfie stick Four Mountain Hardwear Wicked Lite short-sleeved T-shirts 11” MacBook Air laptop Columbia Sportswear rain shell iPhone 6 (and iPhone 4) with an international phone plan Marmot down jacket iPod nano and headphones Two pairs of ExOfficio field pants SureFire Fury LED flashlight Three pairs of ExOfficio Give- with rechargeable batteries N-Go boxer underwear Green laser pointer Two long-sleeved ExOfficio BugsAway insect-repelling Yalumi LED headlamp shirts with sun protection Sea to Summit silk sleeping bag Two pairs of SmartWool socks liner Two pairs of cotton Balega socks Set of adapter plugs for the world Birding Without Borders_F.indd 264 7/14/17 10:49 AM Gear for a Big Year • 265 Wildy Adventure anti-leech Antimalarial pills socks First-aid kit Two bandanas Assorted toiletries (comb, Plain black baseball cap lip balm, eye drops, toenail clippers, tweezers, toothbrush, REI Campware spoon toothpaste, floss, aspirin, Israeli water-purification tablets Imodium, sunscreen) Birding Without Borders_F.indd 265 7/14/17 10:49 AM APPENDIX 2 BIG YEAR SNAPSHOT New Unique per per % % Country Days Total New Unique Day Day New Unique Antarctica / Falklands 8 54 54 30 7 4 100% 56% Argentina 12 435 -
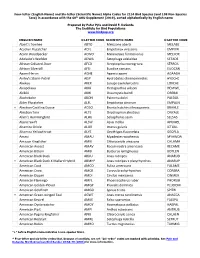
Alpha Codes for 2154 Bird Species (And 108 Non-Species Taxa) in Accordance with the 60Th AOU Supplement (2019), Sorted Alphabetically by English Name
Four-letter (English Name) and Six-letter (Scientific Name) Alpha Codes for 2154 Bird Species (and 108 Non-Species Taxa) in accordance with the 60th AOU Supplement (2019), sorted alphabetically by English name Prepared by Peter Pyle and David F. DeSante The Institute for Bird Populations www.birdpop.org ENGLISH NAME 4-LETTER CODE SCIENTIFIC NAME 6-LETTER CODE Abert's Towhee ABTO Melozone aberti MELABE Acadian Flycatcher ACFL Empidonax virescens EMPVIR Acorn Woodpecker ACWO Melanerpes formicivorus MELFOR Adelaide's Warbler ADWA Setophaga adelaidae SETADE African Collared-Dove AFCD Streptopelia roseogrisea STRROS African Silverbill AFSI Euodice cantans EUOCAN Agami Heron AGHE Agamia agami AGAAGA Ainley's Storm-Petrel AISP Hydrobates cheimomnestes HYDCHE Akekee AKEK Loxops caeruleirostris LOXCAE Akiapolaau AKIA Hemignathus wilsoni HEMWIL Akikiki AKIK Oreomystis bairdi OREBAI Akohekohe AKOH Palmeria dolei PALDOL Alder Flycatcher ALFL Empidonax alnorum EMPALN + Aleutian Cackling Goose ACGO Branta hutchinsii leucopareia BRAHLE Aleutian Tern ALTE Onychoprion aleuticus ONYALE Allen's Hummingbird ALHU Selasphorus sasin SELSAS Alpine Swift ALSW Apus melba APUMEL Altamira Oriole ALOR Icterus gularis ICTGUL Altamira Yellowthroat ALYE Geothlypis flavovelata GEOFLA Amaui AMAU Myadestes woahensis MYAWOA Amazon Kingfisher AMKI Chloroceryle amazona CHLAMA American Avocet AMAV Recurvirostra americana RECAME American Bittern AMBI Botaurus lentiginosus BOTLEN American Black Duck ABDU Anas rubripes ANARUB + American Black Duck X Mallard Hybrid ABMH* Anas -

Seed Dispersal by a Captive Corvid: the Role of the 'Alala¯ (Corvus
Ecological Applications, 22(6), 2012, pp. 1718–1732 Ó 2012 by the Ecological Society of America Seed dispersal by a captive corvid: the role of the ‘Alala¯ (Corvus hawaiiensis) in shaping Hawai‘i’s plant communities 1,4 1 2 1,3 SUSAN CULLINEY, LIBA PEJCHAR, RICHARD SWITZER, AND VIVIANA RUIZ-GUTIERREZ 1Department of Fish, Wildlife, and Conservation Biology, Colorado State University, 1474 Campus Delivery, Fort Collins, Colorado 80523 USA 2Hawai‘i Endangered Bird Conservation Program, San Diego Zoo Institute for Conservation Research, Volcano, Hawaii 96785 USA 3Cornell Laboratory of Ornithology, 159 Sapsucker Woods Road, Cornell University, Ithaca, NewYork 14850 USA Abstract. Species loss can lead to cascading effects on communities, including the disruption of ecological processes such as seed dispersal. The endangered ‘Alala¯ (Corvus hawaiiensis), the largest remaining species of native Hawaiian forest bird, was once common in mesic and dry forests on the Big Island of Hawai‘i, but today it exists solely in captivity. Prior to its extinction in the wild, the ‘Alala¯ may have helped to establish and maintain native Hawaiian forest communities by dispersing seeds of a wide variety of native plants. In the absence of ‘Alala¯, the structure and composition of Hawai‘i’s forests may be changing, and some large-fruited plants may be dispersal limited, persisting primarily as ecological anachronisms. We fed captive ‘Alala¯ a variety of native fruits, documented behaviors relating to seed dispersal, and measured the germination success of seeds that passed through the gut of ‘Alala¯ relative to the germination success of seeds in control groups. ‘Alala¯ ate and carried 14 native fruits and provided germination benefits to several species by ingesting their seeds. -

Inner Mongolia Cumulative Bird List Column A
China: Inner Mongolia Cumulative Bird List Column A: total number of days that the species was recorded in 2016 Column B: maximum daily count for that particular species Column C: H = Heard only; (H) = Heard more often than seen Globally threatened species as defined by BirdLife International (2004) Threatened birds of the world 2004 CD-Rom Cambridge, U.K. BirdLife International are identified as follows: EN = Endangered; VU = Vulnerable; NT = Near- threatened. A B C Ruddy Shelduck 2 3 Tadorna ferruginea Mandarin Duck 1 10 Aix galericulata Gadwall 2 12 Anas strepera Falcated Teal 1 4 Anas falcata Eurasian Wigeon 1 2 Anas penelope Mallard 5 40 Anas platyrhynchos Eastern Spot-billed Duck 3 12 Anas zonorhyncha Eurasian Teal 2 12 Anas crecca Baer's Pochard EN 1 4 Aythya baeri Ferruginous Pochard NT 3 49 Aythya nyroca Tufted Duck 1 1 Aythya fuligula Common Goldeneye 2 7 Bucephala clangula Hazel Grouse 4 14 Tetrastes bonasia Daurian Partridge 1 5 Perdix dauurica Brown Eared Pheasant VU 2 15 Crossoptilon mantchuricum Common Pheasant 8 10 Phasianus colchicus Little Grebe 4 60 Tachybaptus ruficollis Great Crested Grebe 3 15 Podiceps cristatus Eurasian Bittern 3 1 Botaurus stellaris Yellow Bittern 1 1 H Ixobrychus sinensis Black-crowned Night Heron 3 2 Nycticorax nycticorax Chinese Pond Heron 1 1 Ardeola bacchus Grey Heron 3 5 Ardea cinerea Great Egret 1 1 Ardea alba Little Egret 2 8 Egretta garzetta Great Cormorant 1 20 Phalacrocorax carbo Western Osprey 2 1 Pandion haliaetus Black-winged Kite 2 1 Elanus caeruleus ________________________________________________________________________________________________________ WINGS ● 1643 N. Alvernon Way Ste. -

Times to Extinction for Small Populations of Large Birds (Crow/Owl/Hawk/Population Lifetime/Population Size) STUART L
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 10871-10875, November 1993 Population Biology Times to extinction for small populations of large birds (crow/owl/hawk/population lifetime/population size) STUART L. PIMM*, JARED DIAMONDt, TIMOTHY M. REEDt, GARETH J. RUSSELL*, AND JARED VERNER§ *Department of Zoology, University of Tennessee, Knoxville, TN 37996; tDepartment of Physiology, University of California Medical School, Los Angeles, CA 90024-1751; tJoint Nature Conservation Committee, Monkstone House, Peterborough, PE1 1UA, United Kingdom; and §U.S. Department of Agriculture, Forest Service, Pacific Southwest Research Station, Fresno, CA 93710 Contributed by Jared Diamond, August 9, 1993 ABSTRACT A major practical problem in conservation such counts. European islands provide the most extensive biology is to predict the survival times-"lifetimes"-for small existing data set, and hence the best surrogates for 'Alala and populations under alternative proposed management regimes. Spotted Owl populations for the foreseeable future. The data Examples in the United States include the 'Alala (Hawaiian consist of counts of nesting birds tabulated in the annual Crow; Corvus hawaiiensis) and Northern Spotted Owl (Strix reports of bird observatories on small islands around the occidentalis caurina). To guide such decisions, we analyze coasts of Britain and Ireland, and on the German North Sea counts of ail crow, owl, and hawk species in the most complete island of Helgoland. Subsets of these data have been ana- available data set: counts of bird breeding pairs on 14 Euro- lyzed previously (2-9). pean islands censused for 29-66 consecutive years. The data set Our paper expands the previously analyzed data set to yielded 129 records for analysis. -
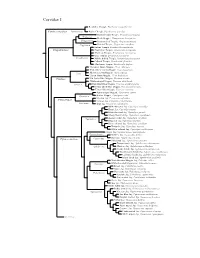
Corvidae Species Tree
Corvidae I Red-billed Chough, Pyrrhocorax pyrrhocorax Pyrrhocoracinae =Pyrrhocorax Alpine Chough, Pyrrhocorax graculus Ratchet-tailed Treepie, Temnurus temnurus Temnurus Black Magpie, Platysmurus leucopterus Platysmurus Racket-tailed Treepie, Crypsirina temia Crypsirina Hooded Treepie, Crypsirina cucullata Rufous Treepie, Dendrocitta vagabunda Crypsirininae ?Sumatran Treepie, Dendrocitta occipitalis ?Bornean Treepie, Dendrocitta cinerascens Gray Treepie, Dendrocitta formosae Dendrocitta ?White-bellied Treepie, Dendrocitta leucogastra Collared Treepie, Dendrocitta frontalis ?Andaman Treepie, Dendrocitta bayleii ?Common Green-Magpie, Cissa chinensis ?Indochinese Green-Magpie, Cissa hypoleuca Cissa ?Bornean Green-Magpie, Cissa jefferyi ?Javan Green-Magpie, Cissa thalassina Cissinae ?Sri Lanka Blue-Magpie, Urocissa ornata ?White-winged Magpie, Urocissa whiteheadi Urocissa Red-billed Blue-Magpie, Urocissa erythroryncha Yellow-billed Blue-Magpie, Urocissa flavirostris Taiwan Blue-Magpie, Urocissa caerulea Azure-winged Magpie, Cyanopica cyanus Cyanopica Iberian Magpie, Cyanopica cooki Siberian Jay, Perisoreus infaustus Perisoreinae Sichuan Jay, Perisoreus internigrans Perisoreus Gray Jay, Perisoreus canadensis White-throated Jay, Cyanolyca mirabilis Dwarf Jay, Cyanolyca nanus Black-throated Jay, Cyanolyca pumilo Silvery-throated Jay, Cyanolyca argentigula Cyanolyca Azure-hooded Jay, Cyanolyca cucullata Beautiful Jay, Cyanolyca pulchra Black-collared Jay, Cyanolyca armillata Turquoise Jay, Cyanolyca turcosa White-collared Jay, Cyanolyca viridicyanus -

Songbird Remix CORVUS CORVUS Contents
Avian Models for 3D Applications by Ken Gilliland 1 Songbird ReMix CORVUS CORVUS Contents Manual Introduction 3 Overview and Use 3 Where to Find your birds and Poses 4 One Folder to Rule them All 4 Physical-based Rendering 5 Posing and Shaping Tips 5 Field Guide Field Guide List 6 Ravens Australian Raven 7 Common Raven 9 White-necked Raven 12 Crows American Crow 14 Eurasian or Western Jackdaw 18 ‘Alala - Hawaiian Crow 21 Rook 25 Credits and Resources 29 Copyrighted 2013-20 by Ken Gilliland www.SongbirdReMix.com Opinions expressed on this booklet are solely that of the author, Ken Gilliland, and may or may not reflect the opinions of the publisher. 2 Songbird ReMix CORVUS CORVUS Introduction Ravens and crows through the ages have represented the darker and sometimes, playful elements in human cultures. In Irish mythology, crows are associated with Morrigan, the goddess of war and death while the Norse believed that ravens carry news to the god Odin about the mortal world. In Hawaiian, Aboriginal and Native American cultures, crows and ravens are believed to be embodiments of ancestor’s spirits or sometimes “The Trickster”; a deity that breaks the rules of the gods or nature, sometimes maliciously but usually with ultimately positive effects. The collective name for a group of crows is a “murder of crows”. Ravens and crows are now considered to be among the world's most intelligent animals. Recent research has found some species capable of not only tool use but also tool construction. The set is located within the Animals : Songbird ReMix folder.