Nothofagus and Pacific Biogeography
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cunninghamia Date of Publication: February 2020 a Journal of Plant Ecology for Eastern Australia
Cunninghamia Date of Publication: February 2020 A journal of plant ecology for eastern Australia ISSN 0727- 9620 (print) • ISSN 2200 - 405X (Online) The Australian paintings of Marianne North, 1880–1881: landscapes ‘doomed shortly to disappear’ John Leslie Dowe Australian Tropical Herbarium, James Cook University, Smithfield, Qld 4878 AUSTRALIA. [email protected] Abstract: The 80 paintings of Australian flora, fauna and landscapes by English artist Marianne North (1830-1890), completed during her travels in 1880–1881, provide a record of the Australian environment rarely presented by artists at that time. In the words of her mentor Sir Joseph Dalton Hooker, director of Kew Gardens, North’s objective was to capture landscapes that were ‘doomed shortly to disappear before the axe and the forest fires, the plough and the flock, or the ever advancing settler or colonist’. In addition to her paintings, North wrote books recollecting her travels, in which she presented her observations and explained the relevance of her paintings, within the principles of a ‘Darwinian vision,’ and inevitable and rapid environmental change. By examining her paintings and writings together, North’s works provide a documented narrative of the state of the Australian environment in the late nineteenth- century, filtered through the themes of personal botanical discovery, colonial expansion and British imperialism. Cunninghamia (2020) 20: 001–033 doi: 10.7751/cunninghamia.2020.20.001 Cunninghamia: a journal of plant ecology for eastern Australia © 2020 Royal Botanic Gardens and Domain Trust www.rbgsyd.nsw.gov.au/science/Scientific_publications/cunninghamia 2 Cunninghamia 20: 2020 John Dowe, Australian paintings of Marianne North, 1880–1881 Introduction The Marianne North Gallery in the Royal Botanic Gardens Kew houses 832 oil paintings which Marianne North (b. -
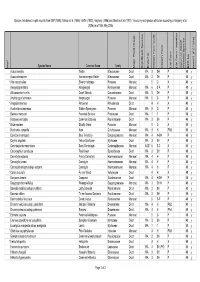
BFS048 Site Species List
Species lists based on plot records from DEP (1996), Gibson et al. (1994), Griffin (1993), Keighery (1996) and Weston et al. (1992). Taxonomy and species attributes according to Keighery et al. (2006) as of 16th May 2005. Species Name Common Name Family Major Plant Group Significant Species Endemic Growth Form Code Growth Form Life Form Life Form - aquatics Common SSCP Wetland Species BFS No kens01 (FCT23a) Wd? Acacia sessilis Wattle Mimosaceae Dicot WA 3 SH P 48 y Acacia stenoptera Narrow-winged Wattle Mimosaceae Dicot WA 3 SH P 48 y * Aira caryophyllea Silvery Hairgrass Poaceae Monocot 5 G A 48 y Alexgeorgea nitens Alexgeorgea Restionaceae Monocot WA 6 S-R P 48 y Allocasuarina humilis Dwarf Sheoak Casuarinaceae Dicot WA 3 SH P 48 y Amphipogon turbinatus Amphipogon Poaceae Monocot WA 5 G P 48 y * Anagallis arvensis Pimpernel Primulaceae Dicot 4 H A 48 y Austrostipa compressa Golden Speargrass Poaceae Monocot WA 5 G P 48 y Banksia menziesii Firewood Banksia Proteaceae Dicot WA 1 T P 48 y Bossiaea eriocarpa Common Bossiaea Papilionaceae Dicot WA 3 SH P 48 y * Briza maxima Blowfly Grass Poaceae Monocot 5 G A 48 y Burchardia congesta Kara Colchicaceae Monocot WA 4 H PAB 48 y Calectasia narragara Blue Tinsel Lily Dasypogonaceae Monocot WA 4 H-SH P 48 y Calytrix angulata Yellow Starflower Myrtaceae Dicot WA 3 SH P 48 y Centrolepis drummondiana Sand Centrolepis Centrolepidaceae Monocot AUST 6 S-C A 48 y Conostephium pendulum Pearlflower Epacridaceae Dicot WA 3 SH P 48 y Conostylis aculeata Prickly Conostylis Haemodoraceae Monocot WA 4 H P 48 y Conostylis juncea Conostylis Haemodoraceae Monocot WA 4 H P 48 y Conostylis setigera subsp. -

Nuytsia the Journal of the Western Australian Herbarium 26: 111–125 Published Online 3 November 2015
R.L. Barrett et al., Seven new species of Haemodorum (Haemodoraceae) 111 Nuytsia The journal of the Western Australian Herbarium 26: 111–125 Published online 3 November 2015 Seven new species of Haemodorum (Haemodoraceae) from the Kimberley region of Western Australia Russell L. Barrett1,2,3,5,6, Stephen D. Hopper4, Terry D. Macfarlane2 and Matthew D. Barrett1,2,3 ¹Botanic Gardens and Parks Authority, Kings Park and Botanic Garden, West Perth, Western Australia 6005 2Western Australian Herbarium, Department of Parks and Wildlife, Locked Bag 104, Bentley Delivery Centre, Western Australia 6983 3School of Plant Biology, Faculty of Science, The University of Western Australia, Crawley, Western Australia 6009 4Centre of Excellence in Natural Resource Management and School of Plant Biology, The University of Western Australia, Albany, Western Australia 6330 5Current address: Australian National Herbarium, Centre for Australian National Biodiversity Research, National Research Collections Australia, CSIRO, GPO Box 1600, Canberra, Australian Capital Territory 2601 6Corresponding author, email: [email protected] Abstract Barrett, R.L., Hopper, S.D., Macfarlane, T.D. and Barrett, M.D. Seven new species of Haemodorum (Haemodoraceae) from the Kimberley region of Western Australia. Nuytsia 26: 111–125 (2015). Haemodorum basalticum R.L.Barrett, Hopper & T.Macfarlane, H. capitatum R.L.Barrett & Hopper, H. condensatum Hopper & R.L.Barrett, H. griseofuscum R.L.Barrett, M.D.Barrett & Hopper, H. interrex R.L.Barrett & M.D.Barrett, H. macfarlanei R.L.Barrett and H. thedae R.L.Barrett are described as new species. Haemodorum basalticum and H. macfarlanei are restricted to the Mitchell Plateau, H. capitatum is restricted to pindan sands in the Dampier Botanical District, H. -

Supporting Information Appendix Pliocene Reversal of Late Neogene
1 Supporting Information Appendix 2 Pliocene reversal of late Neogene aridification 3 4 J.M.K. Sniderman, J. Woodhead, J. Hellstrom, G.J. Jordan, R.N. Drysdale, J.J. Tyler, N. 5 Porch 6 7 8 SUPPLEMENTARY MATERIALS AND METHODS 9 10 Pollen analysis. We attempted to extract fossil pollen from 81 speleothems collected from 11 16 caves from the Western Australian portion of the Nullarbor Plain. Nullarbor speleothems 12 and caves are essentially “fossil” features that appear to have been preserved by very slow 13 rates of landscape change in a semi-arid landscape. Sample collection targeted fallen, well 14 preserved speleothems in multiple caves. U-Pb dates of these speleothems (Table S3) 15 ranged from late Miocene (8.19 Ma) to Middle Pleistocene (0.41 Ma), with an average age 16 of 4.11 Ma. 17 Fossil pollen typically is present in speleothems in very low concentrations, so pollen 18 processing techniques were developed to minimize contamination by modern pollen (1), but 19 also to maximize recovery, to accommodate the highly variable organic matter content of the 20 speleothems, and to remove a clay- to fine silt-sized mineral fraction present in many 21 samples, which was resistant to cold HF and which can become electrostatically attracted to 22 pollen grains, inhibiting their identification. Stalagmite and flowstone samples of 30-200 g 23 mass were first cut on a diamond rock saw in order to remove any obviously porous material. 24 All subsequent physical and chemical processes were carried out within a HEPA-filtered 25 exhausting clean air cabinet in an ISO Class 7 clean room. -

Kew Science Publications for the Academic Year 2017–18
KEW SCIENCE PUBLICATIONS FOR THE ACADEMIC YEAR 2017–18 FOR THE ACADEMIC Kew Science Publications kew.org For the academic year 2017–18 ¥ Z i 9E ' ' . -,i,c-"'.'f'l] Foreword Kew’s mission is to be a global resource in We present these publications under the four plant and fungal knowledge. Kew currently has key questions set out in Kew’s Science Strategy over 300 scientists undertaking collection- 2015–2020: based research and collaborating with more than 400 organisations in over 100 countries What plants and fungi occur to deliver this mission. The knowledge obtained 1 on Earth and how is this from this research is disseminated in a number diversity distributed? p2 of different ways from annual reports (e.g. stateoftheworldsplants.org) and web-based What drivers and processes portals (e.g. plantsoftheworldonline.org) to 2 underpin global plant and academic papers. fungal diversity? p32 In the academic year 2017-2018, Kew scientists, in collaboration with numerous What plant and fungal diversity is national and international research partners, 3 under threat and what needs to be published 358 papers in international peer conserved to provide resilience reviewed journals and books. Here we bring to global change? p54 together the abstracts of some of these papers. Due to space constraints we have Which plants and fungi contribute to included only those which are led by a Kew 4 important ecosystem services, scientist; a full list of publications, however, can sustainable livelihoods and natural be found at kew.org/publications capital and how do we manage them? p72 * Indicates Kew staff or research associate authors. -

Canberra Bird Notes
canberra ISSN 0314-8211 bird Volume 42 Number 3 December 2017 notes Registered by Australia Post 100001304 CANBERRA ORNITHOLOGISTS GROUP, INC. PO Box 301 Civic Square ACT 2608 2017-18 Committee President Neil Hermes 0413 828 045 Vice-President Vacant Secretary Bill Graham 0466 874 723 Treasurer Vacant 6231 0147 (h) Member Jenny Bounds Member Sue Lashko Member Lia Battisson Member David McDonald Member Paul Fennell Member A.O. (Nick) Nicholls Member Steve Read Email contacts General inquiries [email protected] President [email protected] Canberra Bird Notes [email protected]/[email protected] COG Database Inquiries [email protected] COG Membership [email protected] COG Web Discussion List [email protected] Conservation [email protected] Gang-gang Newsletter [email protected] GBS Coordinator [email protected] Publications for sale [email protected] Unusual bird reports [email protected] Website [email protected] Woodland Project [email protected] Other COG contacts Jenny Bounds Conservation Field Trips Sue Lashko 6251 4485 (h) COG Membership Sandra Henderson 6231 0303 (h) Canberra Bird Notes Editor Michael Lenz 6249 1109 (h) Assistant Editor Kevin Windle 6286 8014 (h) Editor for Annual Bird Report Paul Fennell 6254 1804 (h) Newsletter Editor Sue Lashko, Gail Neumann (SL) 6251 4485 (h) Databases Jaron Bailey 0439 270 835 (a.h.) Garden Bird Survey Duncan McCaskill 6259 1843 (h) Rarities Panel Barbara Allan 6254 6520 (h) Talks Program Organiser Jack Holland 6288 7840 (h) Records Officer Nicki Taws 6251 0303 (h) Website Julian Robinson 6239 6226 (h) Sales Kathy Walter 6241 7639 (h) Waterbird Survey Michael Lenz 6249 1109 (h) Distribution of COG publications Dianne Davey 6254 6324 (h) COG Library Barbara Allan 6254 6520 (h) Please use the General Inquiries email to arrange access to library items or for general enquiries, or contact the Secretary on 0466 874 723. -

Pollination Ecology of Trachymene Incisa (Apiaceae
CHAPTER 1 INTRODUCTION Photograph: newly opened Trachymene incisa umbel Introduction 1-2 For each pollinator bee is in fact a little cupid that creates hot romances between plants thought to be stupid. Each bee can be considered a matchmaking agency that arranges introductions for a nominal nectar fee. From “Cupid” by Paul D. Haemig, http://www.ecology.info/cupid.html POLLINATION ECOLOGY Mutualistic interactions are among the most widespread interactions in nature, but also the least well understood compared to other interactions such as predation (Ollerton 2006). Mutualisms are relationships between species that result in reciprocal benefits. Biotic pollination, the transfer of pollen by animals from the anther to the stigma of flowers, is a mostly mutualistic interaction (see Renner 2006 for exceptions), with pollinators usually receiving pollen and/or nectar from plants in return for their pollinating service. Animal pollinators have been important since the angiosperms evolved, with evidence indicating that early members of the angiosperms were insect pollinated (Crane et al. 1995). Over 91% of the 240 000 angiosperm species worldwide are animal-pollinated (Buchmann and Nabhan 1996), distributed throughout approximately 93% of angiosperm genera (Renner and Ricklefs 1995). With vertebrates pollinating only 0.51% of species, insects are responsible for pollinating over 90% of angiosperm species (Buchmann and Nabhan 1996). The four largest orders of insects, Coleoptera, Hymenoptera, Lepidoptera and Diptera, include species that are known to pollinate flowering plants (Proctor et al. 1996). The total number of pollinator species is unknown, although estimates vary between 130 000 and Introduction 1-3 300 000 (Shepherd et al. -

Geophagy: the Earth Eaters of South-Western Australia
GEOPHAGY: THE EARTH-EATERS OF LOWER SOUTHWESTERN AUSTRALIA By Ken Macintyre and Barb Dobson May 2017 This paper was originally written in 2007 based on materials derived from our consultations with Nyungar Elders from south western Australia between 1992 -2007 and extensive archival research at the Battye Library and on-line sources. www.anthropologyfromtheshed.com 1 ABSTRACT Geophagy or the eating of an earthy substance (such as clay) was practised by the Minang Nyungar of southwestern Australia in the King George Sound region prior to and during European colonisation. Earth from termite mounds was added to the processed swollen stems of Haemodorum species (also known as ‘blood roots’ due to their bright red colour, see Plate 1) either as a food additive or a food within its own right. This paper explores, using ethno-historical and anthropological sources, possible explanations for the indigenous cultural practice of geophagy in the extreme southern region of Western Australia. The red bulbous stems of Haemodorum spicatum known locally as meerne in the King George Sound/ Albany area were described by the early Western recorders (such as Nind 1831 and Collie 1834) as the ‘staple’ food of the traditional inhabitants. However, this root vegetable formed only a part of a much broader range of dietary foods that included protein and fat-rich fish, kangaroo, emu, wallaby, possum, birds, bandicoot and bardi. Animal, bird, fish, reptile, plant and insect foods were seasonally exploited depending on their life cycle stage, depending on their enriched fat, protein or carbohydrate content or other nutritional requirements. April was considered a time of plenty when favourite resource foods included fish, frogs, turtles, bandicoot and quinine (processed Macrozamia seedcoat, known as by-yu in the Swan River region. -

A Potential New Cutflower for Australia - Haemodorum Coccineum
A Potential New Cutflower For Australia - Haemodorum Coccineum A report for the Rural Industries Research and Development Corporation by Margaret Johnston and Alenna McMah September 2006 RIRDC Publication No 06/087 RIRDC Project No UQ-117A © 2006 Rural Industries Research and Development Corporation. All rights reserved. ISBN 1 74151 350 2 ISSN 1440-6845 Haemodorum coccineum production in south-east Queensland Publication No. 06/087 Project No. UQ117A The information contained in this publication is intended for general use to assist public knowledge and discussion and to help improve the development of sustainable industries. The information should not be relied upon for the purpose of a particular matter. Specialist and/or appropriate legal advice should be obtained before any action or decision is taken on the basis of any material in this document. The Commonwealth of Australia, Rural Industries Research and Development Corporation, the authors or contributors do not assume liability of any kind whatsoever resulting from any person's use or reliance upon the content of this document. This publication is copyright. However, RIRDC encourages wide dissemination of its research, providing the Corporation is clearly acknowledged. For any other enquiries concerning reproduction, contact the Publications Manager on phone 02 6272 3186. Researcher Contact Details Dr Margaret Johnston Mrs Alenna McMah Centre for Native Floriculture Boomajarril Native Flower Farm Phone: 07 54601240 Phone: 07 54665668 Fax: 07 54601112 Fax: 07 54665668 Email: [email protected] Email: [email protected] In submitting this report, the researcher has agreed to RIRDC publishing this material in its edited form. RIRDC Contact Details Rural Industries Research and Development Corporation Level 2, 15 National Circuit BARTON ACT 2600 PO Box 4776 KINGSTON ACT 2604 Phone: 02 6272 4819 Fax: 02 6272 5877 Email: [email protected]. -

Nomenclatural Notes and New Taxa in the Conostylis Aculeata Group
Nomenclaturalnotes and new taxa in the Conostylis aculeatagroup (Haemodoraceae) By StephenD. Hopper+ Abstract On the basisof detailedfield strLdies of lhe Co osrilisaculeata R.Br. gro.up andan examinationof specimensai themajor Australian berbaria, (i) C. robustt! Dielsis reinstatedas a speciesdistinct from C. acaleara,(ii) confusionconc.erning the ideniityol C. stylidioidesF. Muell. is clarified:this speciesis redescribed a1ldC. prolifercBenth. is r'einslatedas a distinctspecies; and (.iit\C. aculeata ssp.hrevifotu ssp. nov. and C. puLrcif{orasp. nov. are described;both are reslrictedendemics of the westerncoastal region of the SouihWest Botanical Provinceof w.A. A keyto the 7 speciesnow recognized in lhe C. acule.taEtoup is provided. Introduction The Conostylisq(uleatq R.Bt. group consists of a complex of sevenspecies of perennialherbs whicb are restrictedto the westerncoastal plain and nearby plateau regions of south-westernAustralia. The group forms part of the sectior Cor?orryl/.'(Bentham 1873; Geerinck 1969), and is characterizedby leaves with marginal spines or setae,numerous ovules borne all over the placentalsurface, and a basicchromosome number of x : 8. The taxonomy of this speciescomplex has been notably problematical in previonssystematic studies of the genus. Severalauthors have commented on difficultiesin delimiting taxa (Bentham 1873; Ewart 1906; Domin l9l2; Green 1960). Indeed,herbarium studies leave the impressionofa polymorphic but intergradingassemblage ofpopulations wjth few fornrsthat are consistently distinct throughout their geographicalrange. Elsewhere,the present author has shown that both hybridization and ecologically-correlatedintraspecific variation occur in speciesofthe C. aculeqta group near Dawesville on the Swan Coastal Plain (Hopper 1977). It was there denonstrated that while the identification of specimensfrom this region could be difficult in the herbarium, critical lield observatiorlsenabled the satisfactoryplacement of individuals into morphologically definable species and/or their hybrids. -

On the Flora of Australia
L'IBRARY'OF THE GRAY HERBARIUM HARVARD UNIVERSITY. BOUGHT. THE FLORA OF AUSTRALIA, ITS ORIGIN, AFFINITIES, AND DISTRIBUTION; BEING AN TO THE FLORA OF TASMANIA. BY JOSEPH DALTON HOOKER, M.D., F.R.S., L.S., & G.S.; LATE BOTANIST TO THE ANTARCTIC EXPEDITION. LONDON : LOVELL REEVE, HENRIETTA STREET, COVENT GARDEN. r^/f'ORElGN&ENGLISH' <^ . 1859. i^\BOOKSELLERS^.- PR 2G 1.912 Gray Herbarium Harvard University ON THE FLORA OF AUSTRALIA ITS ORIGIN, AFFINITIES, AND DISTRIBUTION. I I / ON THE FLORA OF AUSTRALIA, ITS ORIGIN, AFFINITIES, AND DISTRIBUTION; BEIKG AN TO THE FLORA OF TASMANIA. BY JOSEPH DALTON HOOKER, M.D., F.R.S., L.S., & G.S.; LATE BOTANIST TO THE ANTARCTIC EXPEDITION. Reprinted from the JJotany of the Antarctic Expedition, Part III., Flora of Tasmania, Vol. I. LONDON : LOVELL REEVE, HENRIETTA STREET, COVENT GARDEN. 1859. PRINTED BY JOHN EDWARD TAYLOR, LITTLE QUEEN STREET, LINCOLN'S INN FIELDS. CONTENTS OF THE INTRODUCTORY ESSAY. § i. Preliminary Remarks. PAGE Sources of Information, published and unpublished, materials, collections, etc i Object of arranging them to discuss the Origin, Peculiarities, and Distribution of the Vegetation of Australia, and to regard them in relation to the views of Darwin and others, on the Creation of Species .... iii^ § 2. On the General Phenomena of Variation in the Vegetable Kingdom. All plants more or less variable ; rate, extent, and nature of variability ; differences of amount and degree in different natural groups of plants v Parallelism of features of variability in different groups of individuals (varieties, species, genera, etc.), and in wild and cultivated plants vii Variation a centrifugal force ; the tendency in the progeny of varieties being to depart further from their original types, not to revert to them viii Effects of cross-impregnation and hybridization ultimately favourable to permanence of specific character x Darwin's Theory of Natural Selection ; — its effects on variable organisms under varying conditions is to give a temporary stability to races, species, genera, etc xi § 3. -

Conservation Advice Conostylis Micrantha Small Flowered Conostylis
THREATENED SPECIES SCIENTIFIC COMMITTEE Established under the Environment Protection and Biodiversity Conservation Act 1999 The Minister’s delegate approved this conservation advice on 01/04/2016. Conservation Advice Conostylis micrantha small flowered conostylis Conservation Status Conostylis micrantha (small flowered conostylis) is listed as Endangered under the Environment Protection and Biodiversity Conservation Act 1999 (Cwlth) (EPBC Act). The species is eligible for listing as prior to the commencement of the EPBC Act, it was listed as Endangered under Schedule 1 of the Endangered Species Protection Act 1992 (Cwlth). The main factors that are the cause of the species being eligible for listing in the Endangered category are the effects of wide scale land clearing which has resulted in the loss of most areas of suitable habitat (Stack & Chant 2005). The remaining 10 known populations are small and threatened by low levels of weed invasion and grazing by rabbits (Stack & Chant 2005). Description Small flowered conostylis is a small, tufted perennial herb to 30 cm in diameter with yellow- cream flowers that turn a brick red colour with age. The leaves are 13 to 24 cm long and circular in cross-section, with a few spreading, white hairs 3 to 9 mm long on the lower margins. The flowers are held on stems 5 to 13 cm long, with a hairy papery bract 3 to 8 mm long halfway up the stem (Hopper et al., 1987). The tubular flower is 5 to 7.5 mm long, and divides into six lobes that are cream inside and golden yellow outside (Brown et al., 1998).