Gene-Level Heritability Analysis Explains the Polygenic Architecture of Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Single Cell Derived Clonal Analysis of Human Glioblastoma Links
SUPPLEMENTARY INFORMATION: Single cell derived clonal analysis of human glioblastoma links functional and genomic heterogeneity ! Mona Meyer*, Jüri Reimand*, Xiaoyang Lan, Renee Head, Xueming Zhu, Michelle Kushida, Jane Bayani, Jessica C. Pressey, Anath Lionel, Ian D. Clarke, Michael Cusimano, Jeremy Squire, Stephen Scherer, Mark Bernstein, Melanie A. Woodin, Gary D. Bader**, and Peter B. Dirks**! ! * These authors contributed equally to this work.! ** Correspondence: [email protected] or [email protected]! ! Supplementary information - Meyer, Reimand et al. Supplementary methods" 4" Patient samples and fluorescence activated cell sorting (FACS)! 4! Differentiation! 4! Immunocytochemistry and EdU Imaging! 4! Proliferation! 5! Western blotting ! 5! Temozolomide treatment! 5! NCI drug library screen! 6! Orthotopic injections! 6! Immunohistochemistry on tumor sections! 6! Promoter methylation of MGMT! 6! Fluorescence in situ Hybridization (FISH)! 7! SNP6 microarray analysis and genome segmentation! 7! Calling copy number alterations! 8! Mapping altered genome segments to genes! 8! Recurrently altered genes with clonal variability! 9! Global analyses of copy number alterations! 9! Phylogenetic analysis of copy number alterations! 10! Microarray analysis! 10! Gene expression differences of TMZ resistant and sensitive clones of GBM-482! 10! Reverse transcription-PCR analyses! 11! Tumor subtype analysis of TMZ-sensitive and resistant clones! 11! Pathway analysis of gene expression in the TMZ-sensitive clone of GBM-482! 11! Supplementary figures and tables" 13" "2 Supplementary information - Meyer, Reimand et al. Table S1: Individual clones from all patient tumors are tumorigenic. ! 14! Fig. S1: clonal tumorigenicity.! 15! Fig. S2: clonal heterogeneity of EGFR and PTEN expression.! 20! Fig. S3: clonal heterogeneity of proliferation.! 21! Fig. -

Genetic Characterization of Greek Population Isolates Reveals Strong Genetic Drift at Missense and Trait-Associated Variants
ARTICLE Received 22 Apr 2014 | Accepted 22 Sep 2014 | Published 6 Nov 2014 DOI: 10.1038/ncomms6345 OPEN Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants Kalliope Panoutsopoulou1,*, Konstantinos Hatzikotoulas1,*, Dionysia Kiara Xifara2,3, Vincenza Colonna4, Aliki-Eleni Farmaki5, Graham R.S. Ritchie1,6, Lorraine Southam1,2, Arthur Gilly1, Ioanna Tachmazidou1, Segun Fatumo1,7,8, Angela Matchan1, Nigel W. Rayner1,2,9, Ioanna Ntalla5,10, Massimo Mezzavilla1,11, Yuan Chen1, Chrysoula Kiagiadaki12, Eleni Zengini13,14, Vasiliki Mamakou13,15, Antonis Athanasiadis16, Margarita Giannakopoulou17, Vassiliki-Eirini Kariakli5, Rebecca N. Nsubuga18, Alex Karabarinde18, Manjinder Sandhu1,8, Gil McVean2, Chris Tyler-Smith1, Emmanouil Tsafantakis12, Maria Karaleftheri16, Yali Xue1, George Dedoussis5 & Eleftheria Zeggini1 Isolated populations are emerging as a powerful study design in the search for low-frequency and rare variant associations with complex phenotypes. Here we genotype 2,296 samples from two isolated Greek populations, the Pomak villages (HELIC-Pomak) in the North of Greece and the Mylopotamos villages (HELIC-MANOLIS) in Crete. We compare their genomic characteristics to the general Greek population and establish them as genetic isolates. In the MANOLIS cohort, we observe an enrichment of missense variants among the variants that have drifted up in frequency by more than fivefold. In the Pomak cohort, we find novel associations at variants on chr11p15.4 showing large allele frequency increases (from 0.2% in the general Greek population to 4.6% in the isolate) with haematological traits, for example, with mean corpuscular volume (rs7116019, P ¼ 2.3 Â 10 À 26). We replicate this association in a second set of Pomak samples (combined P ¼ 2.0 Â 10 À 36). -

Table S3. RAE Analysis of Well-Differentiated Liposarcoma
Table S3. RAE analysis of well-differentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes Genes Amp 1 145009467 145122002 112536 0.097 21.8 2 PRKAB2,PDIA3P Amp 1 145224467 146188434 963968 0.029 23.6 10 CHD1L,BCL9,ACP6,GJA5,GJA8,GPR89B,GPR89C,PDZK1P1,RP11-94I2.2,NBPF11 Amp 1 147475854 148412469 936616 0.034 23.6 20 PPIAL4A,FCGR1A,HIST2H2BF,HIST2H3D,HIST2H2AA4,HIST2H2AA3,HIST2H3A,HIST2H3C,HIST2H4B,HIST2H4A,HIST2H2BE, HIST2H2AC,HIST2H2AB,BOLA1,SV2A,SF3B4,MTMR11,OTUD7B,VPS45,PLEKHO1 Amp 1 148582896 153398462 4815567 1.5E-05 49.1 152 PRPF3,RPRD2,TARS2,ECM1,ADAMTSL4,MCL1,ENSA,GOLPH3L,HORMAD1,CTSS,CTSK,ARNT,SETDB1,LASS2,ANXA9, FAM63A,PRUNE,BNIPL,C1orf56,CDC42SE1,MLLT11,GABPB2,SEMA6C,TNFAIP8L2,LYSMD1,SCNM1,TMOD4,VPS72, PIP5K1A,PSMD4,ZNF687,PI4KB,RFX5,SELENBP1,PSMB4,POGZ,CGN,TUFT1,SNX27,TNRC4,MRPL9,OAZ3,TDRKH,LINGO4, RORC,THEM5,THEM4,S100A10,S100A11,TCHHL1,TCHH,RPTN,HRNR,FLG,FLG2,CRNN,LCE5A,CRCT1,LCE3E,LCE3D,LCE3C,LCE3B, LCE3A,LCE2D,LCE2C,LCE2B,LCE2A,LCE4A,KPRP,LCE1F,LCE1E,LCE1D,LCE1C,LCE1B,LCE1A,SMCP,IVL,SPRR4,SPRR1A,SPRR3, SPRR1B,SPRR2D,SPRR2A,SPRR2B,SPRR2E,SPRR2F,SPRR2C,SPRR2G,LELP1,LOR,PGLYRP3,PGLYRP4,S100A9,S100A12,S100A8, S100A7A,S100A7L2,S100A7,S100A6,S100A5,S100A4,S100A3,S100A2,S100A16,S100A14,S100A13,S100A1,C1orf77,SNAPIN,ILF2, NPR1,INTS3,SLC27A3,GATAD2B,DENND4B,CRTC2,SLC39A1,CREB3L4,JTB,RAB13,RPS27,NUP210L,TPM3,C1orf189,C1orf43,UBAP2L,HAX1, AQP10,ATP8B2,IL6R,SHE,TDRD10,UBE2Q1,CHRNB2,ADAR,KCNN3,PMVK,PBXIP1,PYGO2,SHC1,CKS1B,FLAD1,LENEP,ZBTB7B,DCST2, DCST1,ADAM15,EFNA4,EFNA3,EFNA1,RAG1AP1,DPM3 Amp 1 -

Output Results of CLIME (Clustering by Inferred Models of Evolution)
Output results of CLIME (CLustering by Inferred Models of Evolution) Dataset: Num of genes in input gene set: 4 Total number of genes: 20834 Prediction LLR threshold: 0 The CLIME PDF output two sections: 1) Overview of Evolutionarily Conserved Modules (ECMs) Top panel shows the predefined species tree. Bottom panel shows the partition of input genes into Evolutionary Conserved Modules (ECMs), ordered by ECM strength (shown at right), and separated by horizontal lines. Each row show one gene, where the phylogenetic profile indicates presence (blue) or absence (gray) of homologs in each species (column). Gene symbols are shown at left. Gray color indicates that the gene is a paralog to a higher scoring gene within the same ECM (based on BLASTP E < 1e-3). 2) Details of each ECM and its expansion ECM+ Top panel shows the inferred evolutionary history on the predefined species tree. Branch color shows the gain event (blue) and loss events (red color, with brighter color indicating higher confidence in loss). Branches before the gain or after a loss are shown in gray. Bottom panel shows the input genes that are within the ECM (blue/white rows) as well as all genes in the expanded ECM+ (green/gray rows). The ECM+ includes genes likely to have arisen under the inferred model of evolution relative to a background model, and scored using a log likelihood ratio (LLR). PG indicates "paralog group" and are labeled alphabetically (i.e., A, B). The first gene within each paralog group is shown in black color. All other genes sharing sequence similarity (BLAST E < 1e-3) are assigned to the same PG label and displayed in gray. -

Mean Depth 1000 More Than100×
A B ×20 ×50 ×100 100% 1000 800 90% 600 80% 400 Mean depth 70% 200 0 60% 50% Mean coverage 40% PBL002_T PBL003_T PBL004_T PBL005_T PBL006_T PBL007_T PBL008_T PBL009_T PBL010_T PBL001_P 30% 20% 10% 0% PBL002_T PBL003_T PBL004_T PBL005_T PBL006_T PBL007_T PBL008_T PBL009_T PBL010_T PBL001_P Figure S1. Depths and coverages of targeted deep sequencing. A. Depths of targeted deep sequencing on 10 PBL cases (mean depth = 677). B. Coverages of the targeted sequences analyzed at the indicated depths; 98.8% of the targeted regions were sequenced at a depth more than 100×. A B ×2 ×10 ×20 250 100% 200 90% 150 80% 100 Mean depth 50 70% 0 60% 50% PBL002_T PBL004_T PBL007_T PBL008_T PBL010_T PBL001_P PBL001_N PBL002_N PBL004_N PBL007_N PBL008_N PBL010_N PBL001_M1 PBL001_M2 PBL001_M3 PBL001_M4 PBL001_M5 PBL001_M6 Mean coverage 40% 30% 20% 10% 0% PBL002_T PBL004_T PBL007_T PBL008_T PBL010_T PBL001_P PBL001_N PBL002_N PBL004_N PBL007_N PBL008_N PBL010_N PBL001_M1 PBL001_M2 PBL001_M3 PBL001_M4 PBL001_M5 PBL001_M6 C D 14 80 12 70 10 60 50 8 40 6 30 4 Number of mutations Number of mutations 20 2 10 0 0 PBL002_T PBL004_T PBL007_T PBL008_T PBL010_T PBL001_P PBL001_P PBL001_M1 PBL001_M2 PBL001_M3 PBL001_M4 PBL001_M5 PBL001_M6 Nonsynonymous SNV Stopgain SNV Nonframeshift indels Frameshift indels Splice site Figure S2. Sequencing coverages and numbers of mutations detected by WES. A. Depths of WES on tumor and normal DNA samples from 6 cases (mean depth = 124 for tumor samples and 141 for normal samples). B. Coverages of the exome sequences; 97.5% of the exome sequences were analyzed at a depth more than 20×. C-D. -
Two-Dimensional Enrichment Analysis for Mining High-Level Imaging Genetic Associations
Brain Informatics (2017) 4:27–37 DOI 10.1007/s40708-016-0052-4 Two-dimensional enrichment analysis for mining high-level imaging genetic associations Xiaohui Yao . Jingwen Yan . Sungeun Kim . Kwangsik Nho . Shannon L. Risacher . Mark Inlow . Jason H. Moore . Andrew J. Saykin . Li Shen Received: 21 January 2016 / Accepted: 29 April 2016 / Published online: 13 May 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Enrichment analysis has been widely applied in expression data from Allen Human Brain Atlas and the genome-wide association studies, where gene sets imaging genetics data from Alzheimer’s Disease Neu- corresponding to biological pathways are examined for roimaging Initiative as test beds, we present an IGEA significant associations with a phenotype to help increase framework and conduct a proof-of-concept study. This statistical power and improve biological interpretation. In empirical study identifies 25 significant high-level two-di- this work, we expand the scope of enrichment analysis into mensional imaging genetics modules. Many of these brain imaging genetics, an emerging field that studies how modules are relevant to a variety of neurobiological path- genetic variation influences brain structure and function ways or neurodegenerative diseases, showing the promise measured by neuroimaging quantitative traits (QT). Given of the proposal framework for providing insight into the the high dimensionality of both imaging and genetic data, mechanism of complex diseases. we propose to study Imaging Genetic Enrichment Analysis (IGEA), a new enrichment analysis paradigm that jointly Keywords Imaging genetics Á Enrichment analysis Á considers meaningful gene sets (GS) and brain circuits Genome-wide association study Á Quantitative trait (BC) and examines whether any given GS–BC pair is enriched in a list of gene–QT findings. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

Mining High-Level Brain Imaging Genetic Associations
MINING HIGH-LEVEL BRAIN IMAGING GENETIC ASSOCIATIONS Xiaohui Yao Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the School of Informatics and Computing, Indiana University March 2018 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Huanmei Wu, PhD, Chair Doctoral Committee Li Shen, PhD January 16, 2018 Shiaofen Fang, PhD Jingwen Yan, PhD ii c 2018 Xiaohui Yao iii DEDICATION Dedicated to my dear family for all their love and support along the way iv ACKNOWLEDGEMENTS First and foremost I would like to express my deepest gratitude to my advisor, Dr. Li Shen, for guiding and supporting my research during the past five years. He has provided me with great research insights and exceptional enthusiasm as well as encouragement throughout my PhD study. This work would never be materialized without him. I also give my thanks to all the committee members for supporting this thesis work and all the constructive suggestions and feedback, that have been very helpful to keep this work on track and finally make the goal accomplished timely. I would like to express my appreciation to all my colleagues and professors from Center for Neuroimaging: Dr. Andrew J. Saykin, Dr. Shannon L. Risacher, Dr. Kwangsik Nho, and many others, who have provided me very valuable domain exper- tise from neurological, biological and genetic perspectives, as well as many invaluable medical data sources. I have learned considerably from their multi-perspective in- sights into problems. -
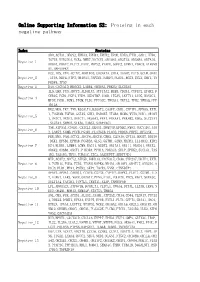
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

A TUMOR SUPPRESSOR-REGULATED CELL CYCLE GENE SIGNATURE IS PROGNOSTIC of RECURRENCE RISK in PROSTATE CANCER Georgescu Et Al
SUPPLEMENTARY MATERIALS FOR: A TUMOR SUPPRESSOR-REGULATED CELL CYCLE GENE SIGNATURE IS PROGNOSTIC OF RECURRENCE RISK IN PROSTATE CANCER Georgescu et al. This PDF file includes: Supplementary Methods Supplementary Discussion Supplementary References Supplementary Figures S1-S11 Supplementary Tables S1-S7 S1 SUPPLEMENTARY METHODS Patient samples for TMEFF2 immunohistochemistry Radical prostatectomy histopathology records from the Department of Pathology at the University of Oklahoma Health Sciences Center (OUSHC) were retrospectively examined. De-identified, archived, PCa specimens from Oklahoma patients with localized and metastatic disease, collected between 2005 and 2015, were obtained (n=39). Only pathological T-score and Gleason score clinical data was captured. All the other clinical samples were from publically available datasets described below. Institutional Review Board Approval This study was approved by the institutional review board of the University of Oklahoma Health Sciences Center. The institutional review board issued an expedited review and waived the need for written consent for this study because only archival, de-identified materials were used. Histology and Immunohistochemistry Paraffin embedded prostate biopsies were selected and 5-μm sections were prepared for hematoxilin & eosin (H&E) staining and immunohistochemistry (IHC). IHC was performed according to manufacturer’s protocol using Leica Bond-IIITM Polymer Refine Detection system (DS 9800). Briefly, the slides with the sections were deparaffinized and rehydrated in an automated Multistainer (Leica ST5020, Leica Biosystems, Buffalo Grove, IL) and transferred to the Leica Bond-IIITM for antigen retrieval at 100°C (20-40 minutes). Endogenous peroxidase was blocked using peroxidase-blocking reagent, followed by S2 incubation with the TMEFF2 antibody (Sigma; 1:150 dilution) for 60 minutes and then post-primary IgG-linker and/or Poly-HRP IgG reagents. -

Functional Analysis of Genome-Wide Dataset from 17000 Individuals Identifies Multiple Candidate Malaria Resistance Genes Enriched in Malaria Pathogenic Pathways
medRxiv preprint doi: https://doi.org/10.1101/2020.08.15.20175471; this version posted August 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Functional analysis of genome-wide dataset from 17000 individuals identifies multiple candidate malaria resistance genes enriched in malaria pathogenic pathways Delesa Damena1, Francis Agamah1, Peter O. Kimathi1, Ntumba E. Kabongo1, Hundaol Girma1, Wonderful T. Choga1, Lemu Golassa3 and Emile R. Chimusa1,2* 1Division of Human Genetics, Department of Pathology, University of Cape Town, Private Bag, Rondebosch, 7700, Cape Town, South Africa. 2Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Private Bag, Rondebosch, 7700, Cape Town, South Africa. 3Addis Ababa University, Aklilu Lema Institute of Pathobiology, P.O.Box 1176, Addis Ababa, Ethiopia Abstract Recent genome-wide association studies (GWASs) of severe malaria have identified several association variants. However, much about the underlying biological functions are yet to be discovered. Here, we systematically predicted plausible candidate genes and pathways from functional analysis of severe malaria resistance GWAS summary statistics(N=17,000) meta- analyzed across eleven populations in malaria endemic regions. We applied positional mapping, expression quantitative trait locus (eQTL), chromatin interaction mapping and gene-based association analyses to identify candidate severe malaria resistance genes. We performed network and pathway analyses to investigate their shared biological functions. We further applied rare variant analysis to raw GWAS datasets (N=11,000) of three malaria endemic populations including Kenya, Malawi and Gambia and performed various population genetic structures of the identified genes in the three populations and global populations.